

MOG CNS Autoimmunity and MOGAD
- PMID: 38996203
- PMCID: PMC11256982
- DOI: 10.1212/NXI.0000000000200275
MOG CNS Autoimmunity and MOGAD
Abstract
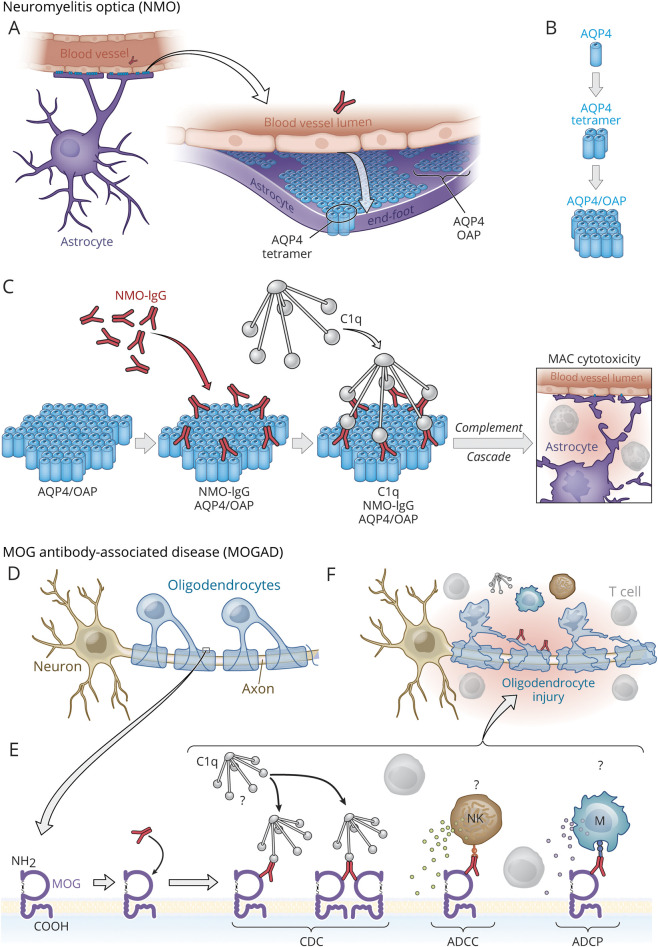
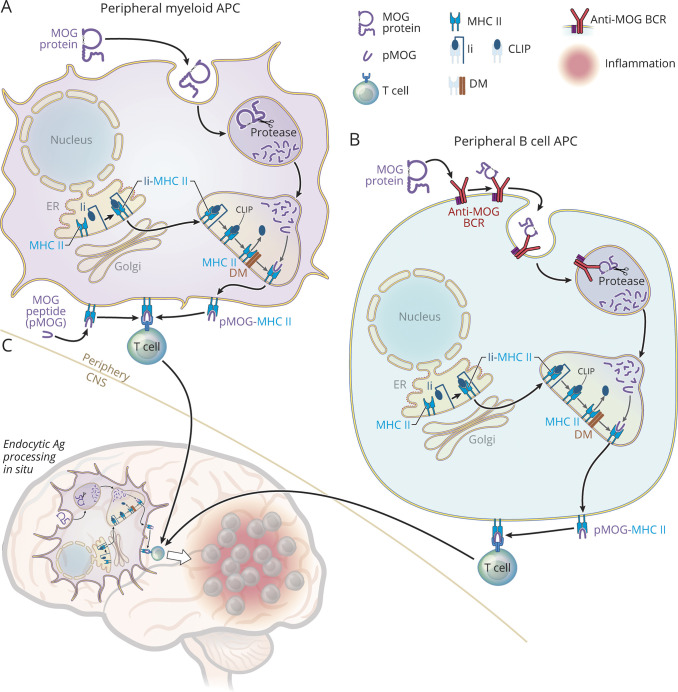
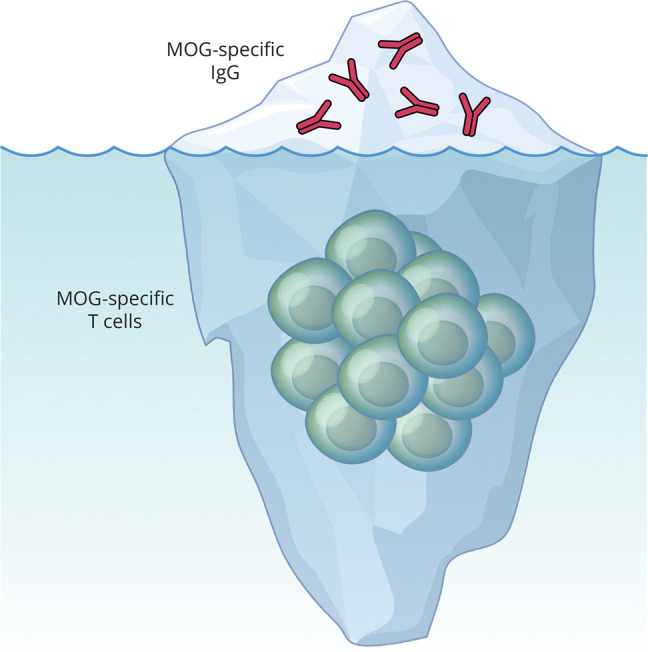
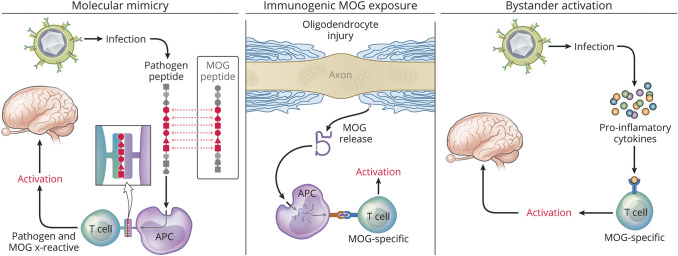
At one time considered a possible form of neuromyelitis optica (NMO) spectrum disorder (NMOSD), it is now accepted that myelin oligodendrocyte glycoprotein (MOG) antibody (Ab)-associated disorder (MOGAD) is a distinct entity from either NMO or multiple sclerosis (MS) and represents a broad spectrum of clinical phenotypes. Whereas Abs targeting aquaporin-4 (AQP4) in NMO are pathogenic, the extent that anti-MOG Abs contribute to CNS damage in MOGAD is unclear. Both AQP4-specific Abs in NMO and MOG-specific Abs in MOGAD are predominantly IgG1, a T cell-dependent immunoglobulin (Ig) subclass. Key insights in neuroimmunology and MOGAD pathogenesis have been learned from MOG experimental autoimmune encephalomyelitis (EAE), described 2 decades before the term MOGAD was introduced. MOG-specific T cells are required in MOG EAE, and while anti-MOG Abs can exacerbate EAE and CNS demyelination, those Abs are neither necessary nor sufficient to cause EAE. Knowledge regarding the spectrum of MOGAD clinical and radiologic presentations is advancing rapidly, yet our grasp of MOGAD pathogenesis is incomplete. Understanding both the humoral and cellular immunology of MOGAD has implications for diagnosis, treatment, and prognosis.
Conflict of interest statement
The authors report no relevant disclosures. Go to
Figures
References
-
- Kerlero de Rosbo N, Mendel I, Ben-Nun A. Chronic relapsing experimental autoimmune encephalomyelitis with a delayed onset and an atypical clinical course, induced in PL/J mice by myelin oligodendrocyte glycoprotein (MOG)-derived peptide: preliminary analysis of MOG T cell epitopes. Eur J Immunol. 1995;25(4):985-993. doi:10.1002/eji.1830250419 - DOI - PubMed