

Benchmarking and integrating human B-cell receptor genomic and antibody proteomic profiling
- PMID: 38997321
- PMCID: PMC11245537
- DOI: 10.1038/s41540-024-00402-z
Benchmarking and integrating human B-cell receptor genomic and antibody proteomic profiling
Abstract
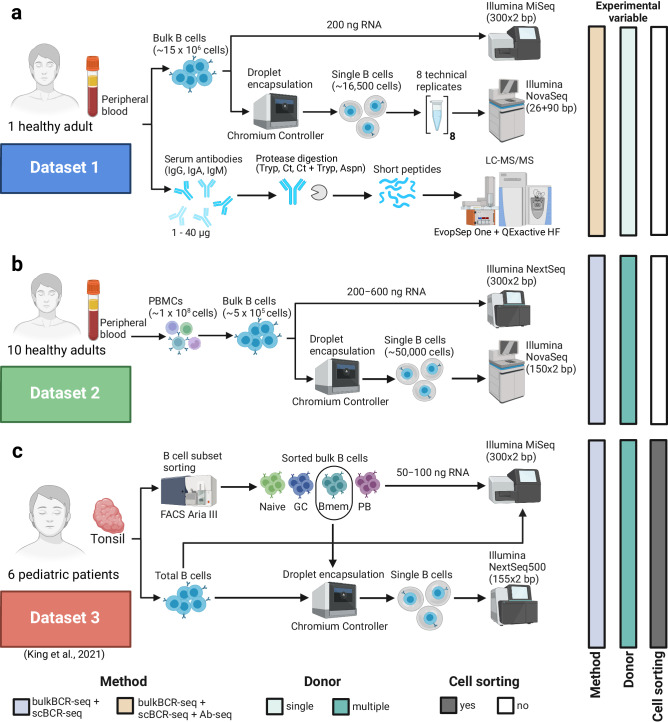
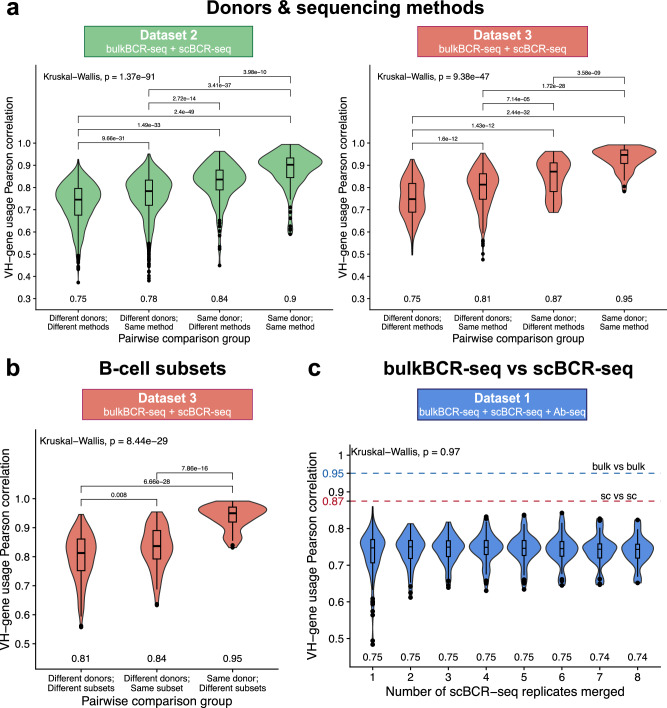
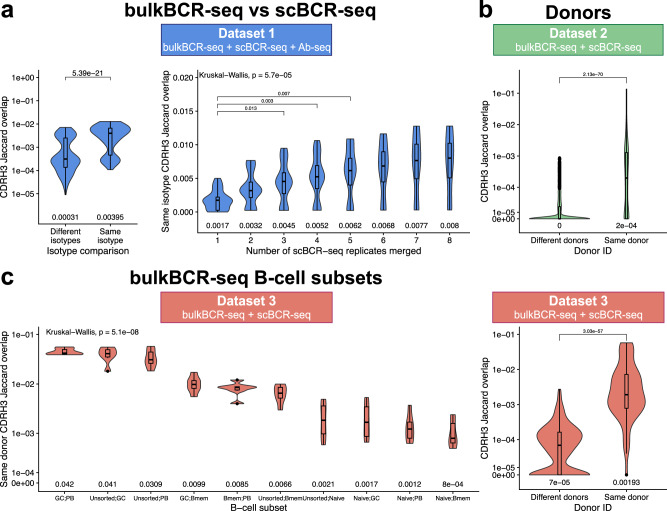
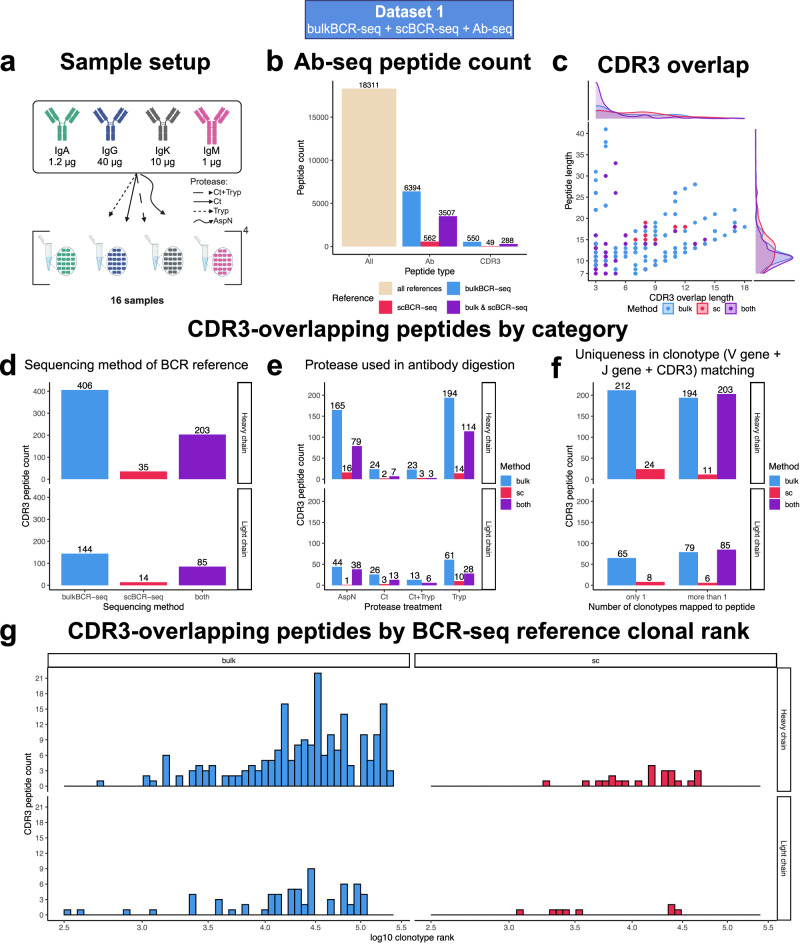
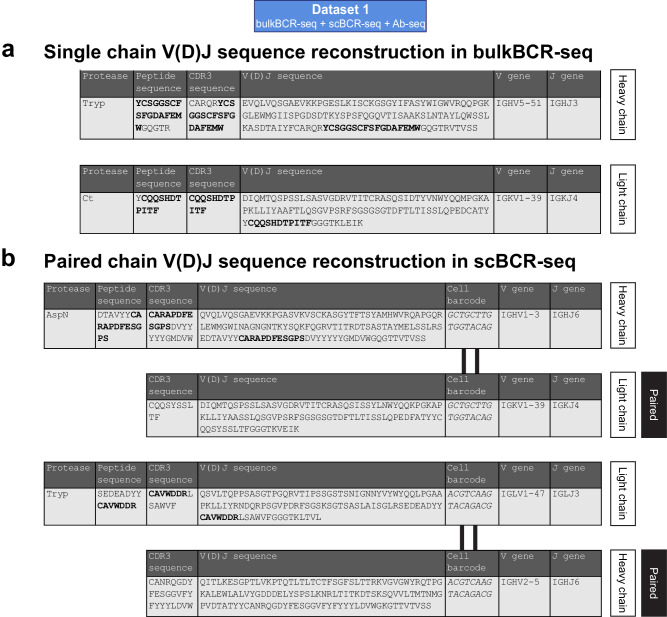
Immunoglobulins (Ig), which exist either as B-cell receptors (BCR) on the surface of B cells or as antibodies when secreted, play a key role in the recognition and response to antigenic threats. The capability to jointly characterize the BCR and antibody repertoire is crucial for understanding human adaptive immunity. From peripheral blood, bulk BCR sequencing (bulkBCR-seq) currently provides the highest sampling depth, single-cell BCR sequencing (scBCR-seq) allows for paired chain characterization, and antibody peptide sequencing by tandem mass spectrometry (Ab-seq) provides information on the composition of secreted antibodies in the serum. Yet, it has not been benchmarked to what extent the datasets generated by these three technologies overlap and complement each other. To address this question, we isolated peripheral blood B cells from healthy human donors and sequenced BCRs at bulk and single-cell levels, in addition to utilizing publicly available sequencing data. Integrated analysis was performed on these datasets, resolved by replicates and across individuals. Simultaneously, serum antibodies were isolated, digested with multiple proteases, and analyzed with Ab-seq. Systems immunology analysis showed high concordance in repertoire features between bulk and scBCR-seq within individuals, especially when replicates were utilized. In addition, Ab-seq identified clonotype-specific peptides using both bulk and scBCR-seq library references, demonstrating the feasibility of combining scBCR-seq and Ab-seq for reconstructing paired-chain Ig sequences from the serum antibody repertoire. Collectively, our work serves as a proof-of-principle for combining bulk sequencing, single-cell sequencing, and mass spectrometry as complementary methods towards capturing humoral immunity in its entirety.
© 2024. The Author(s).
Conflict of interest statement
V.G. declares advisory board positions in aiNET GmbH, Enpicom B.V, Absci, Omniscope, and Diagonal Therapeutics. V.G. is a consultant for Adaptyv Biosystems, Specifica Inc, Roche/Genentech, immunai, Proteinea, LabGenius, and FairJourney Biologics. All other authors declare no competing interests.
Figures

Similar articles
-
Next-generation sequencing and protein mass spectrometry for the comprehensive analysis of human cellular and serum antibody repertoires.Curr Opin Chem Biol. 2015 Feb;24:112-20. doi: 10.1016/j.cbpa.2014.11.007. Epub 2014 Nov 25. Curr Opin Chem Biol. 2015. PMID: 25461729 Review.
-
Capturing the differences between humoral immunity in the normal and tumor environments from repertoire-seq of B-cell receptors using supervised machine learning.BMC Bioinformatics. 2019 May 28;20(1):267. doi: 10.1186/s12859-019-2853-y. BMC Bioinformatics. 2019. PMID: 31138102 Free PMC article.
-
nf-core/airrflow: An adaptive immune receptor repertoire analysis workflow employing the Immcantation framework.PLoS Comput Biol. 2024 Jul 26;20(7):e1012265. doi: 10.1371/journal.pcbi.1012265. eCollection 2024 Jul. PLoS Comput Biol. 2024. PMID: 39058741 Free PMC article.
-
Global characterization of B cell receptor repertoire in COVID-19 patients by single-cell V(D)J sequencing.Brief Bioinform. 2021 Nov 5;22(6):bbab192. doi: 10.1093/bib/bbab192. Brief Bioinform. 2021. PMID: 34015809 Free PMC article.
-
iReceptor: A platform for querying and analyzing antibody/B-cell and T-cell receptor repertoire data across federated repositories.Immunol Rev. 2018 Jul;284(1):24-41. doi: 10.1111/imr.12666. Immunol Rev. 2018. PMID: 29944754 Free PMC article. Review.
Cited by
-
Computational mining of B cell receptor repertoires reveals antigen-specific and convergent responses to Ebola vaccination.Front Immunol. 2024 Jul 8;15:1383753. doi: 10.3389/fimmu.2024.1383753. eCollection 2024. Front Immunol. 2024. PMID: 39040106 Free PMC article. Clinical Trial.
-
Serum proteomics reveals high-affinity and convergent antibodies by tracking SARS-CoV-2 hybrid immunity to emerging variants of concern.Front Immunol. 2025 Feb 25;16:1509888. doi: 10.3389/fimmu.2025.1509888. eCollection 2025. Front Immunol. 2025. PMID: 40070844 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
