

Tumor evolution metrics predict recurrence beyond 10 years in locally advanced prostate cancer
- PMID: 38997466
- PMCID: PMC11424488
- DOI: 10.1038/s43018-024-00787-0
Tumor evolution metrics predict recurrence beyond 10 years in locally advanced prostate cancer
Abstract
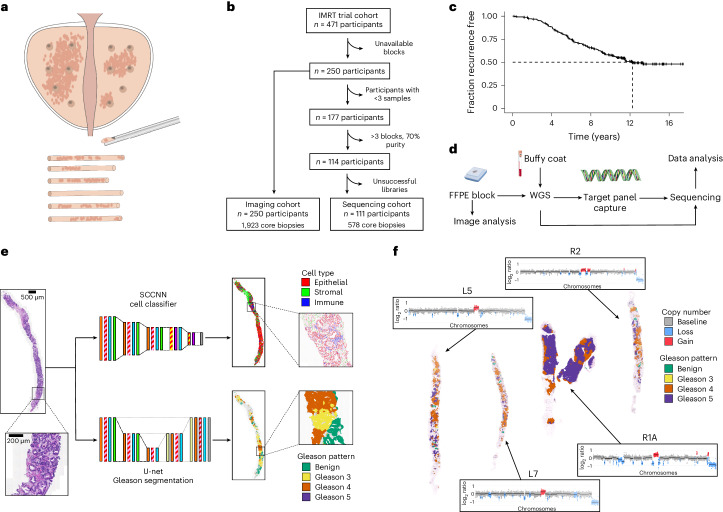
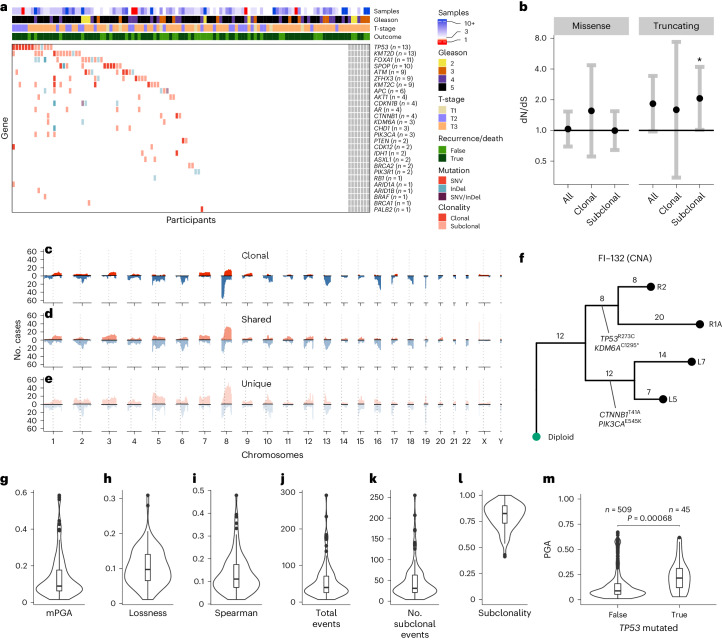
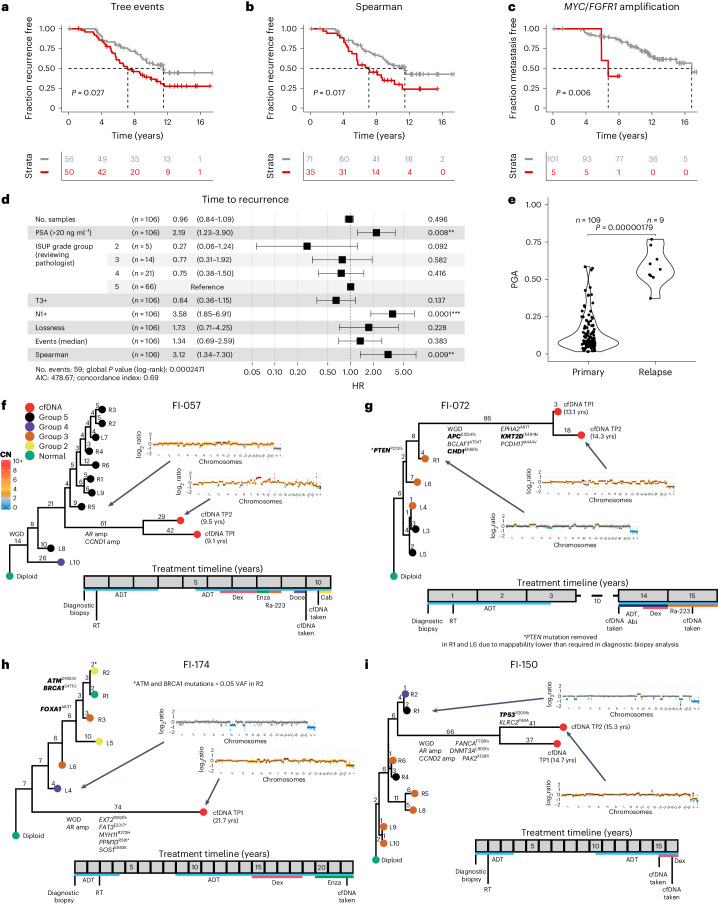
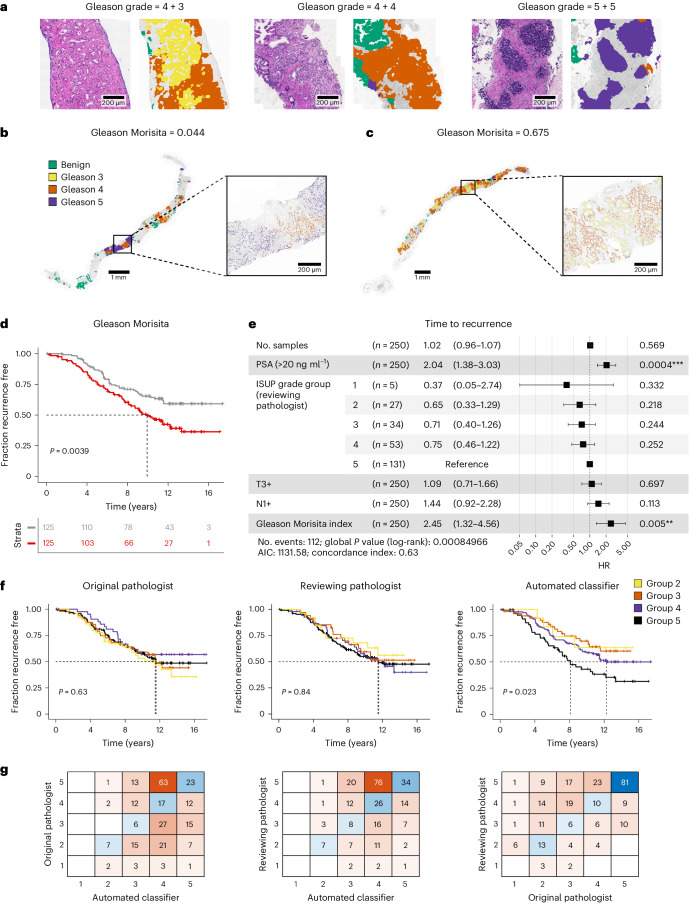
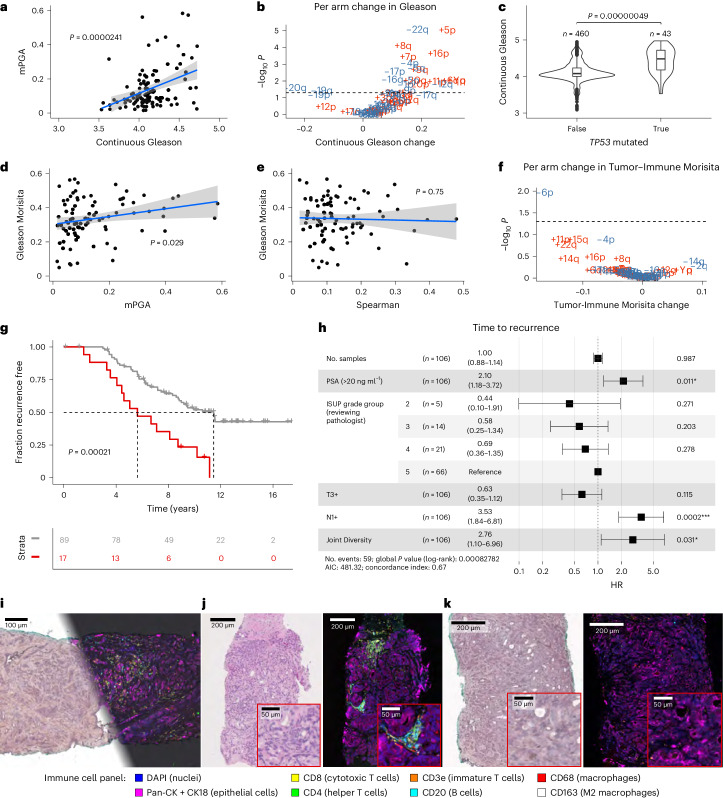
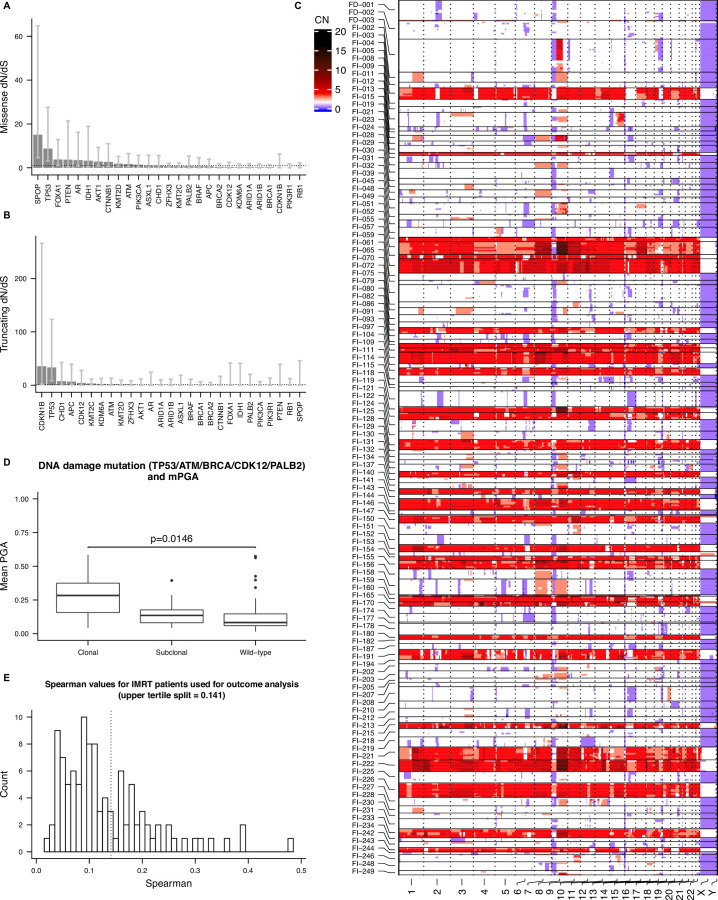
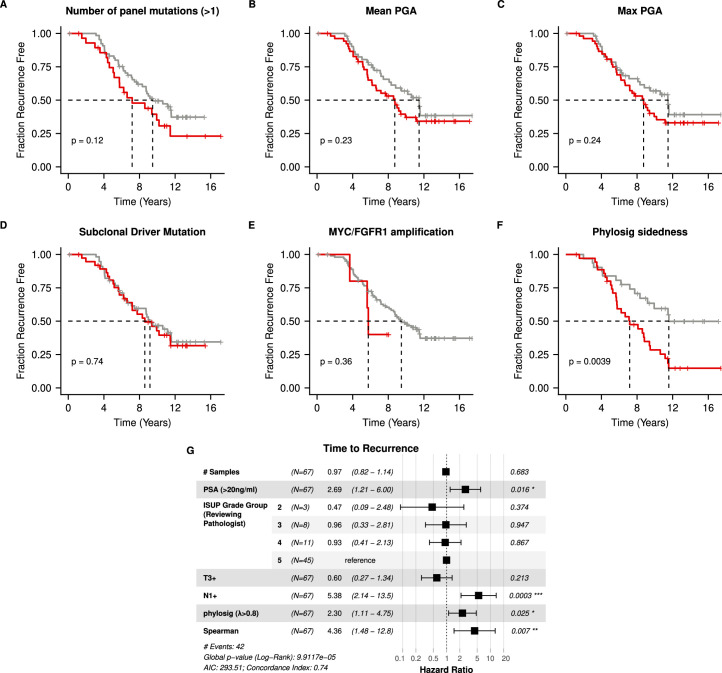
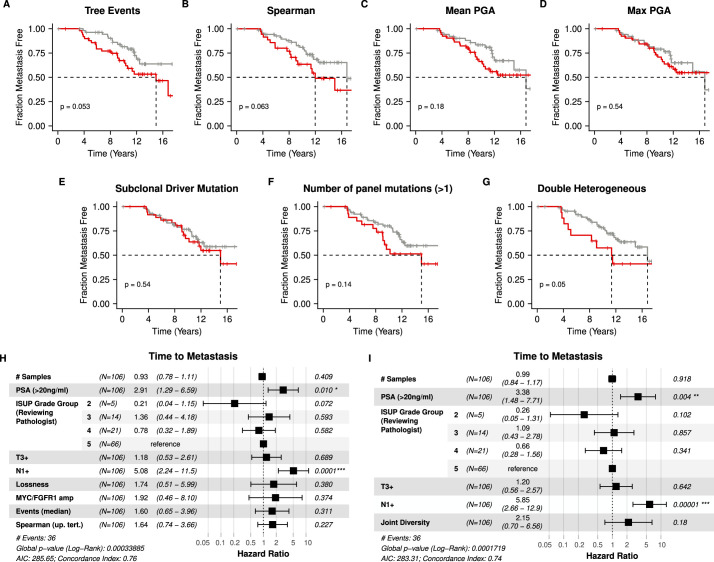
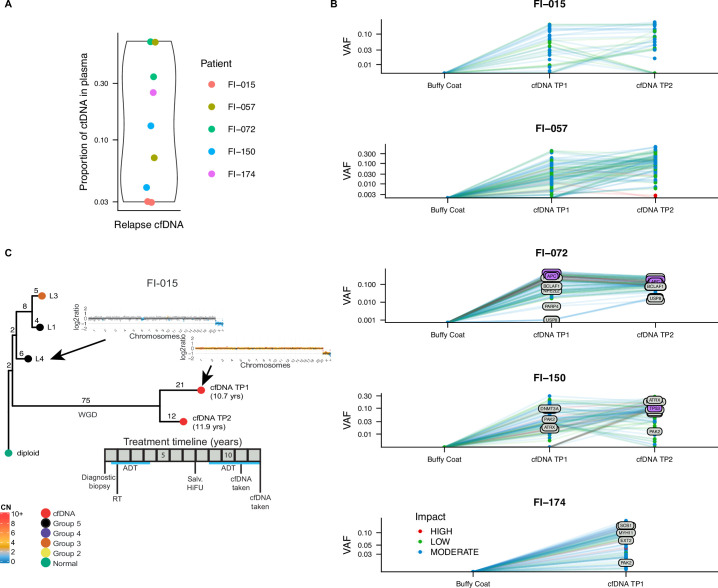
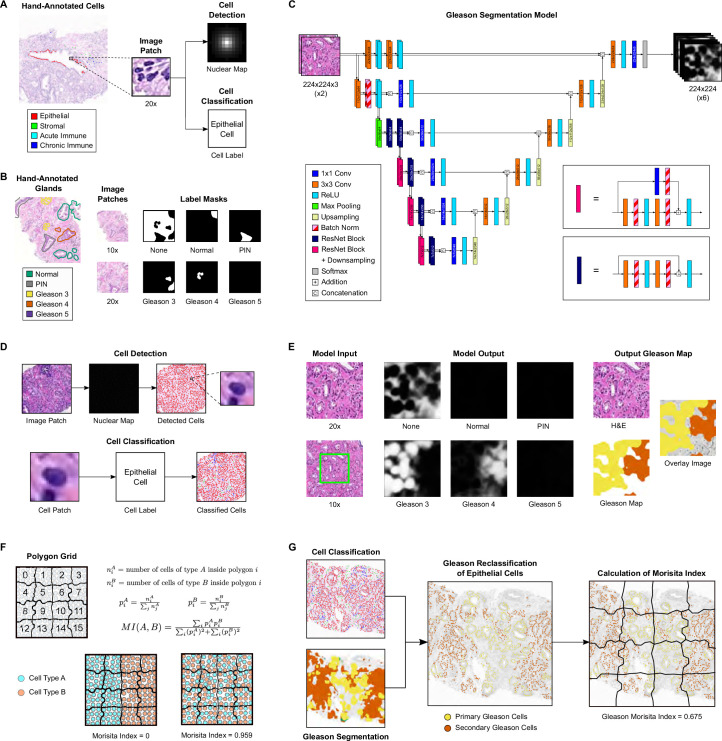
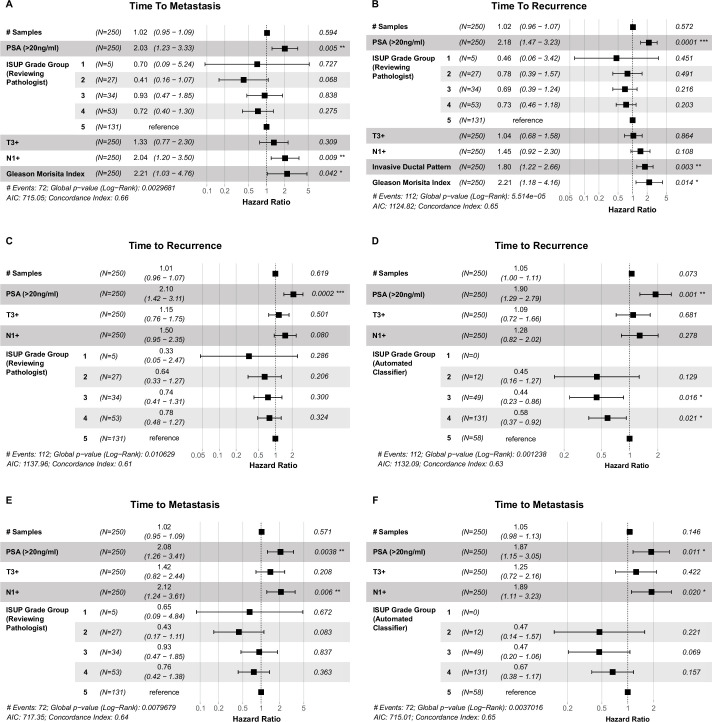
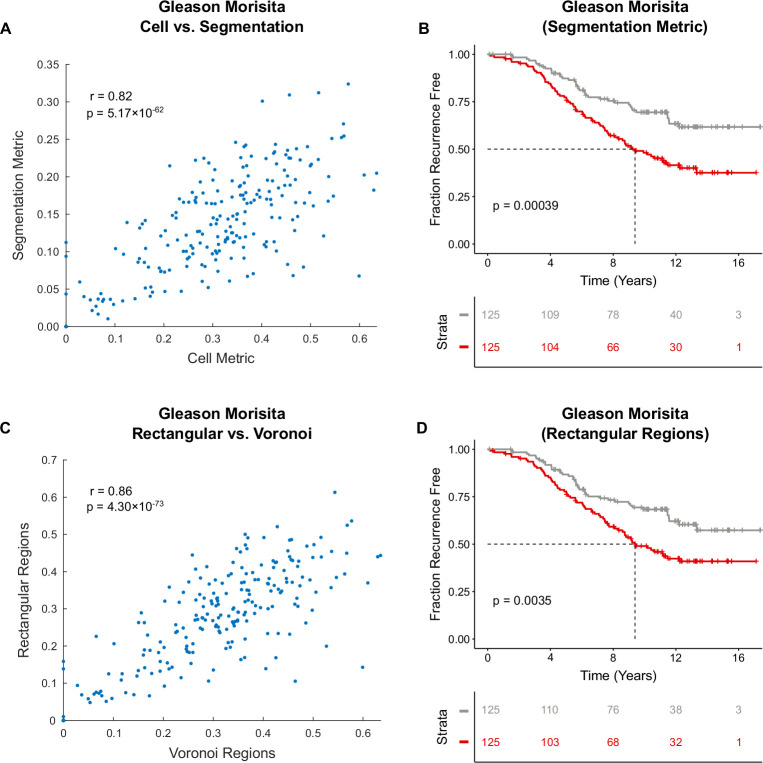
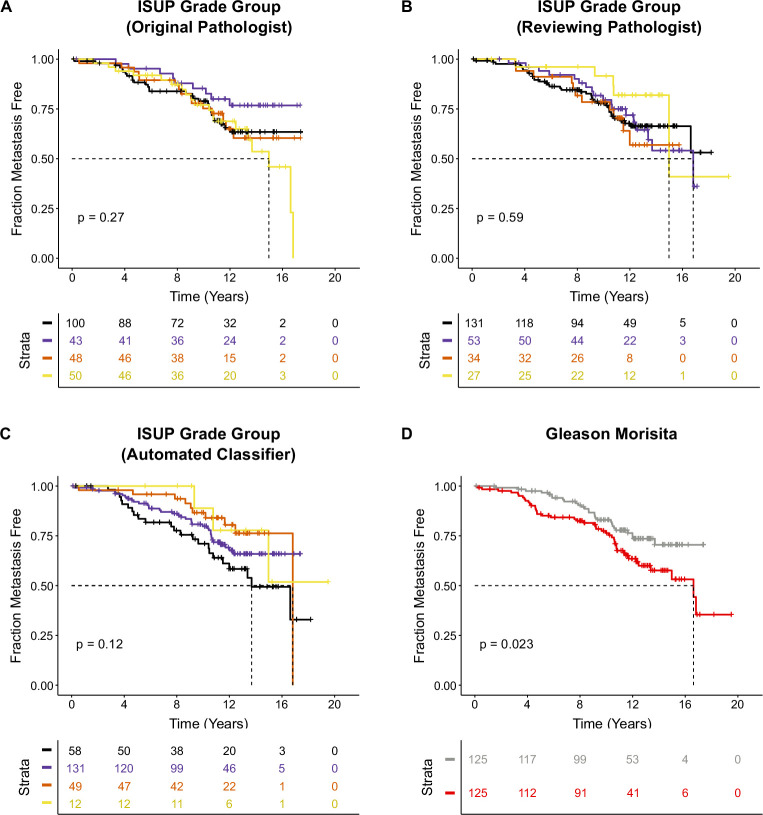
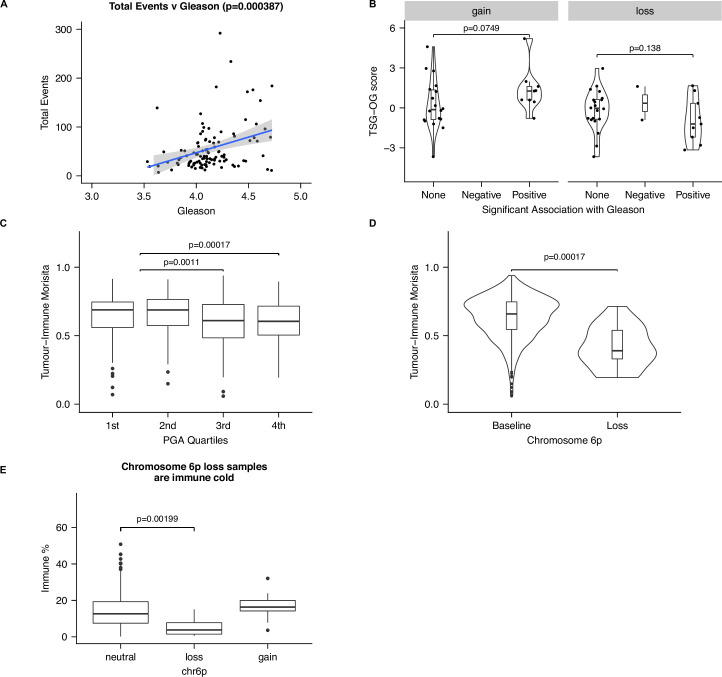
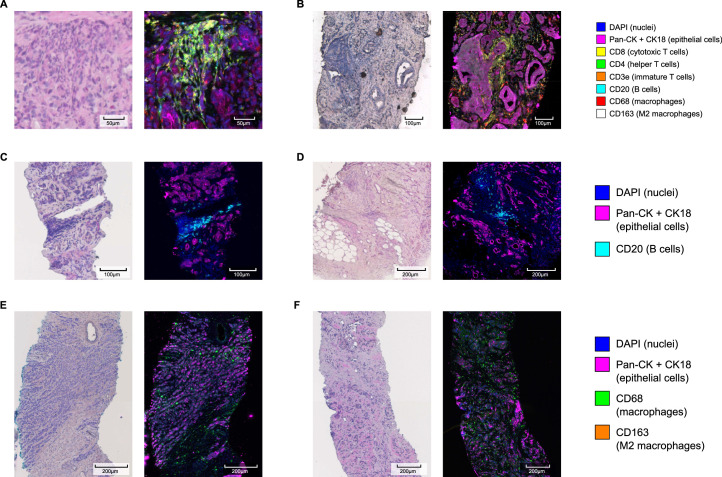
Cancer evolution lays the groundwork for predictive oncology. Testing evolutionary metrics requires quantitative measurements in controlled clinical trials. We mapped genomic intratumor heterogeneity in locally advanced prostate cancer using 642 samples from 114 individuals enrolled in clinical trials with a 12-year median follow-up. We concomitantly assessed morphological heterogeneity using deep learning in 1,923 histological sections from 250 individuals. Genetic and morphological (Gleason) diversity were independent predictors of recurrence (hazard ratio (HR) = 3.12 and 95% confidence interval (95% CI) = 1.34-7.3; HR = 2.24 and 95% CI = 1.28-3.92). Combined, they identified a group with half the median time to recurrence. Spatial segregation of clones was also an independent marker of recurrence (HR = 2.3 and 95% CI = 1.11-4.8). We identified copy number changes associated with Gleason grade and found that chromosome 6p loss correlated with reduced immune infiltration. Matched profiling of relapse, decades after diagnosis, confirmed that genomic instability is a driving force in prostate cancer progression. This study shows that combining genomics with artificial intelligence-aided histopathology leads to the identification of clinical biomarkers of evolution.
Trial registration: ClinicalTrials.gov NCT00946543.
© 2024. The Author(s).
Conflict of interest statement
D.D.’s previous employer, the Institute of Cancer Research, receives royalty income from abiraterone. D.D. receives a share of this income through the Institute of Cancer Research’s Rewards to Discoverer’s Scheme Patent (EP1933709B1), issued for a localization and stabilization device in Europe, Canada and India. D.D. receives honoraria from Janssen Pharmaceuticals. R.E. has the following conflicts of interest to declare: Honoraria from GU-ASCO, Janssen, University of Chicago, Dana Farber Cancer Institute USA as a speaker. Educational honorarium from Bayer and Ipsen, member of external expert committee to Astra Zeneca UK and Member of Active Surveillance Movember Committee. She is a member of the SAB of Our Future Health. She undertakes private practice as a sole trader at The Royal Marsden NHS Foundation Trust and 90 Sloane Street SW1X 9PQ and 280 Kings Road SW3 4NX, London, UK. The other authors declare no competing interests.
Figures
References
-
- Zelic, R. et al. Predicting prostate cancer death with different pretreatment risk stratification tools: a head-to-head comparison in a nationwide cohort study. Eur. Urol.77, 180–188 (2020). - PubMed
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
