

Self-Association and Microhydration of Phenol: Identification of Large-Amplitude Hydrogen Bond Librational Modes
- PMID: 38998964
- PMCID: PMC11243154
- DOI: 10.3390/molecules29133012
Self-Association and Microhydration of Phenol: Identification of Large-Amplitude Hydrogen Bond Librational Modes
Abstract
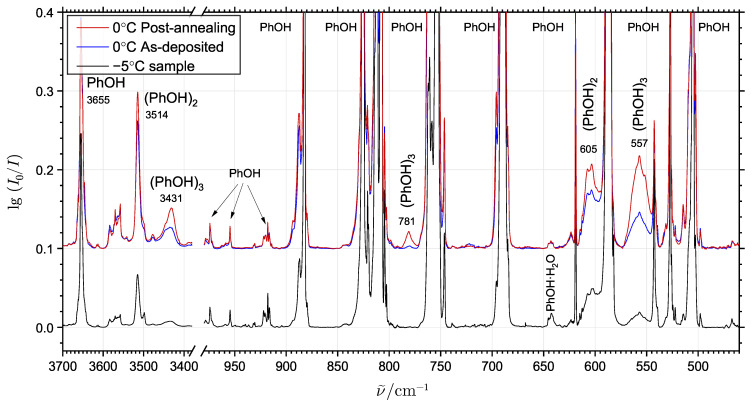
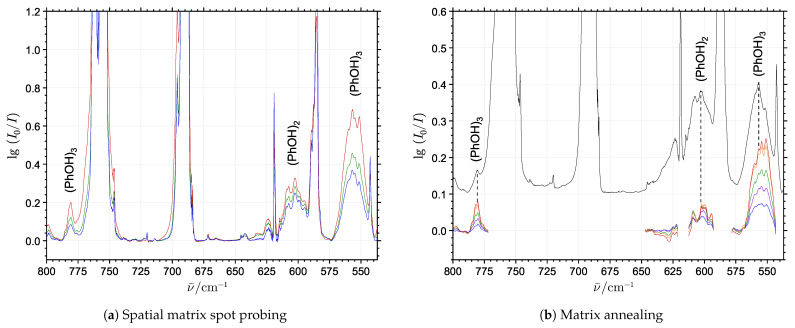
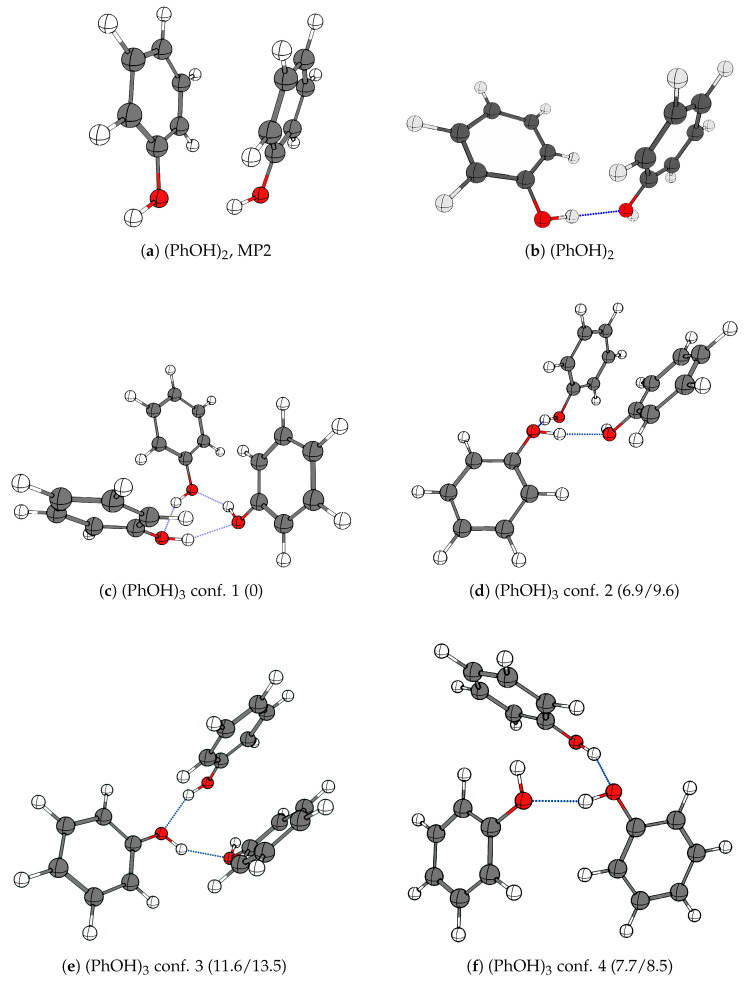
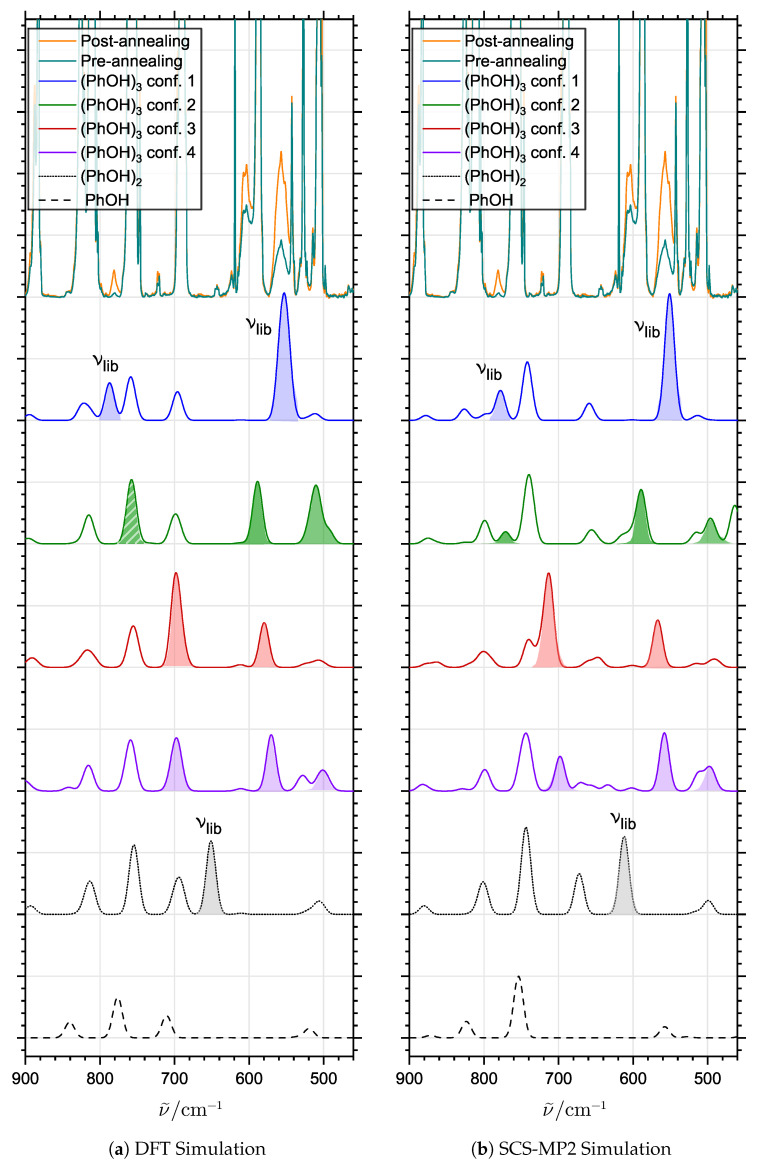
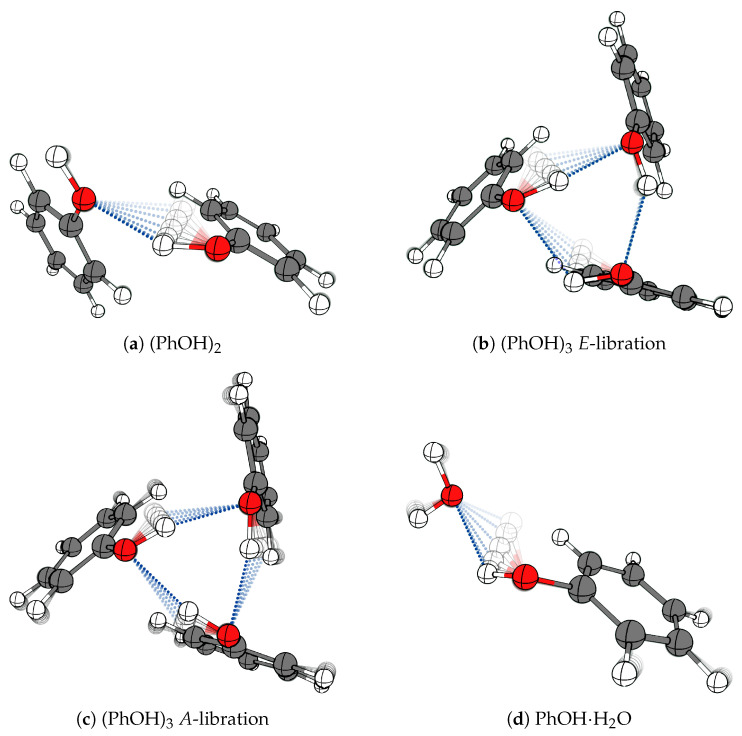
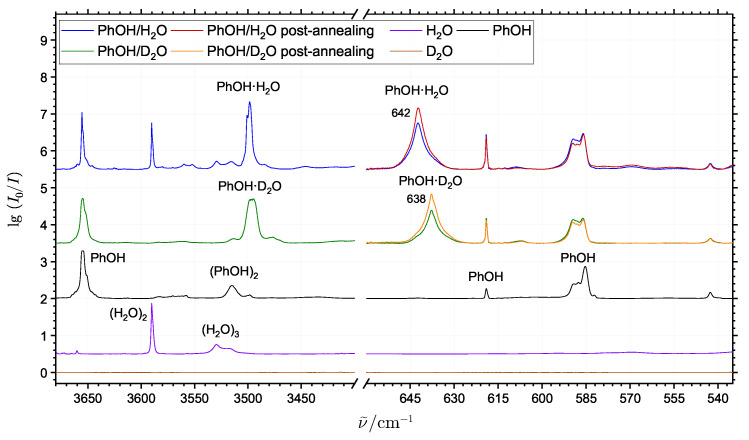
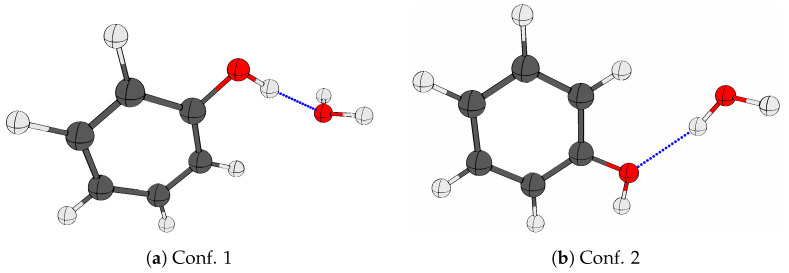
The self-association mechanisms of phenol have represented long-standing challenges to quantum chemical methodologies owing to the competition between strongly directional intermolecular hydrogen bonding, weaker non-directional London dispersion forces and C-H⋯π interactions between the aromatic rings. The present work explores these subtle self-association mechanisms of relevance for biological molecular recognition processes via spectroscopic observations of large-amplitude hydrogen bond librational modes of phenol cluster molecules embedded in inert neon "quantum" matrices complemented by domain-based local pair natural orbital-coupled cluster DLPNO-CCSD(T) theory. The spectral signatures confirm a primarily intermolecular O-H⋯H hydrogen-bonded structure of the phenol dimer strengthened further by cooperative contributions from inter-ring London dispersion forces as supported by DLPNO-based local energy decomposition (LED) predictions. In the same way, the hydrogen bond librational bands observed for the trimeric cluster molecule confirm a pseudo-C3 symmetric cyclic cooperative hydrogen-bonded barrel-like potential energy minimum structure. This structure is vastly different from the sterically favored "chair" conformations observed for aliphatic alcohol cluster molecules of the same size owing to the additional stabilizing London dispersion forces and C-H⋯π interactions between the aromatic rings. The hydrogen bond librational transition observed for the phenol monohydrate finally confirms that phenol acts as a hydrogen bond donor to water in contrast to the hydrogen bond acceptor role observed for aliphatic alcohols.
Keywords: London dispersion forces; hydrogen bonding; large-amplitude librational motion; local energy decomposition; neon matrices; phenol cluster molecules; vibrational spectroscopy.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
LinkOut - more resources
Full Text Sources
