

Genotype-Phenotype Correlation in Neurofibromatosis Type 1: Evidence for a Mild Phenotype Associated with Splicing Variants Leading to In-Frame Skipping of NF1 Exon 24 [19a]
- PMID: 39001468
- PMCID: PMC11240586
- DOI: 10.3390/cancers16132406
Genotype-Phenotype Correlation in Neurofibromatosis Type 1: Evidence for a Mild Phenotype Associated with Splicing Variants Leading to In-Frame Skipping of NF1 Exon 24 [19a]
Abstract
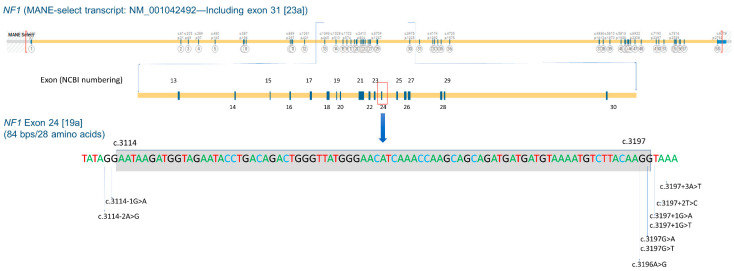
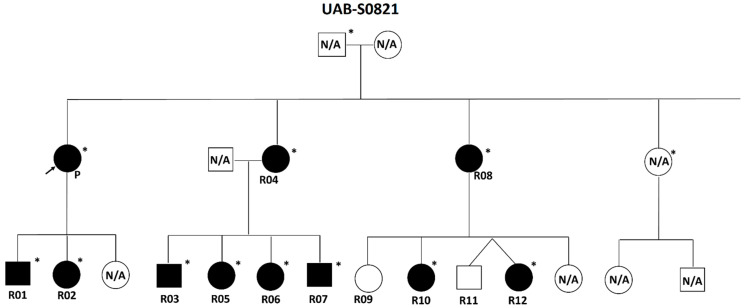
Neurofibromatosis type 1 (NF1) is an autosomal dominant neurocutaneous disorder caused by loss-of-function variants in the NF1 gene. As of 20 November 2023, over 5000 distinct pathogenic or likely pathogenic variants have been reported in public databases. However, only a few NF1 genotype-phenotype correlations have been established so far. In this study, we present findings on 40 individuals with NF1, comprising 26 unrelated probands and 14 affected relatives, who carry one of nine NF1 heterozygous pathogenic splicing variants, all of which result in the in-frame skipping of exon 24 [19a] (NM_000267.3:r.3114_3197del, p.Asn1039_Arg1066del). These variants include c.3114-2A>G, c.3114-1G>A, c.3196A>G, c.3197G>A, c.3197G>T, c.3197+1G>A, c.3197+1G>T, c.3197+2T>C, and c.3197+3A>T. Among individuals with these variants, none exhibit externally visible plexiform neurofibromas, histopathologically confirmed cutaneous or subcutaneous neurofibromas, symptomatic spinal neurofibromas, or symptomatic optic pathway gliomas. The most prevalent, and sometimes sole, clinical feature observed in this cohort is multiple café-au-lait macules, with or without skinfold freckles: 85% and 60.5% of the individuals display six or more café-au-lait macules and freckles, respectively. In comparison to established NF1 genotype-phenotype correlations, these patients demonstrate highly similar clinical presentations to those associated with the NF1 pathogenic variant c.2970_2972del (p.Met992del), known for resulting in the mildest clinical features. Despite the generally mild phenotype, cognitive impairment, developmental delay, and/or learning difficulties are still observed in 33.3% of these patients, suggesting that learning challenges remain a prominent aspect of the phenotypic presentation in these individuals and necessitate specialized care. This newly established genotype-phenotype correlation will assist clinicians in improving the management of patients harboring NF1 exon 24 [19a] skipping variants and provide a new therapeutic target for NF1 treatment.
Keywords: NF1; exon skipping; genotype–phenotype correlation; splicing variant.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Legius E., Messiaen L., Wolkenstein P., Pancza P., Avery R.A., Berman Y., Blakeley J., Babovic-Vuksanovic D., Cunha K.S., Ferner R., et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet. Med. 2021;23:1506–1513. doi: 10.1038/s41436-021-01170-5. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
