

Biallelic NDC1 variants that interfere with ALADIN binding are associated with neuropathy and triple A-like syndrome
- PMID: 39003500
- PMCID: PMC11375137
- DOI: 10.1016/j.xhgg.2024.100327
Biallelic NDC1 variants that interfere with ALADIN binding are associated with neuropathy and triple A-like syndrome
Abstract
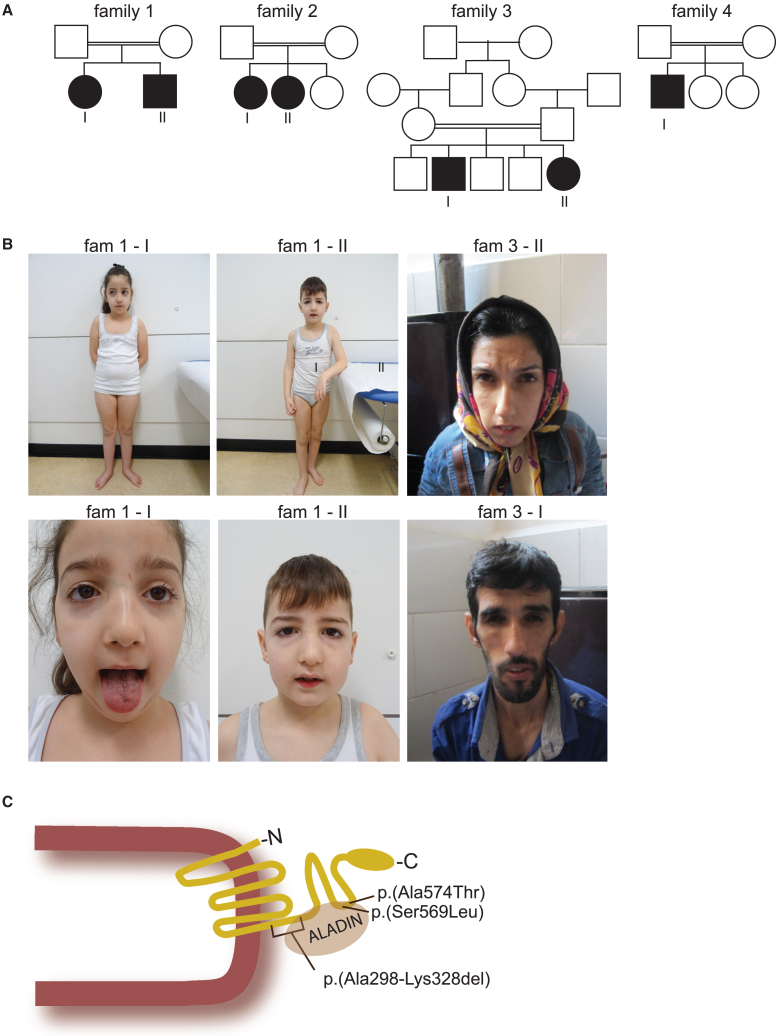
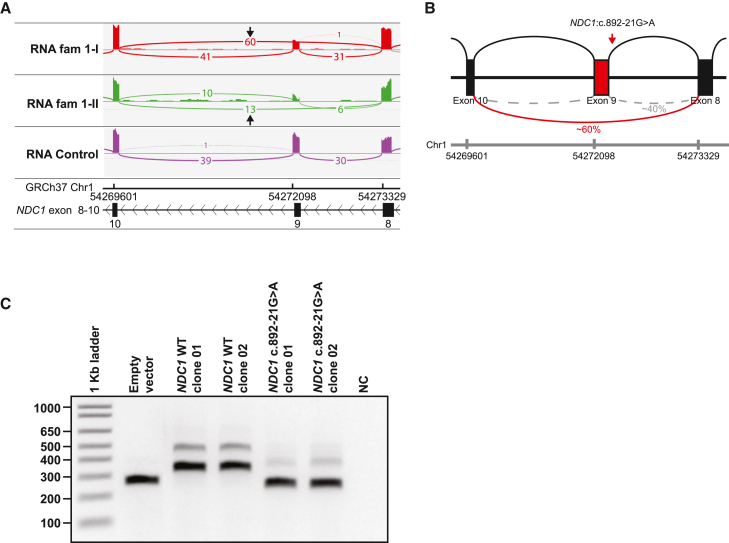
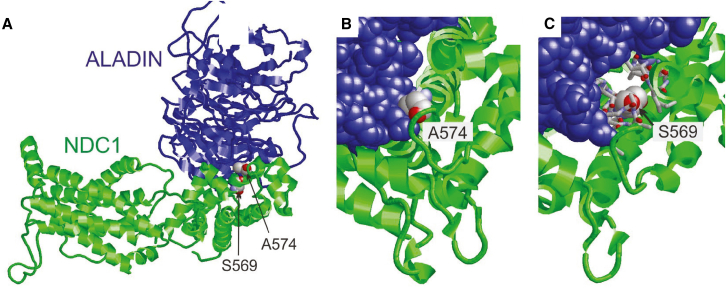
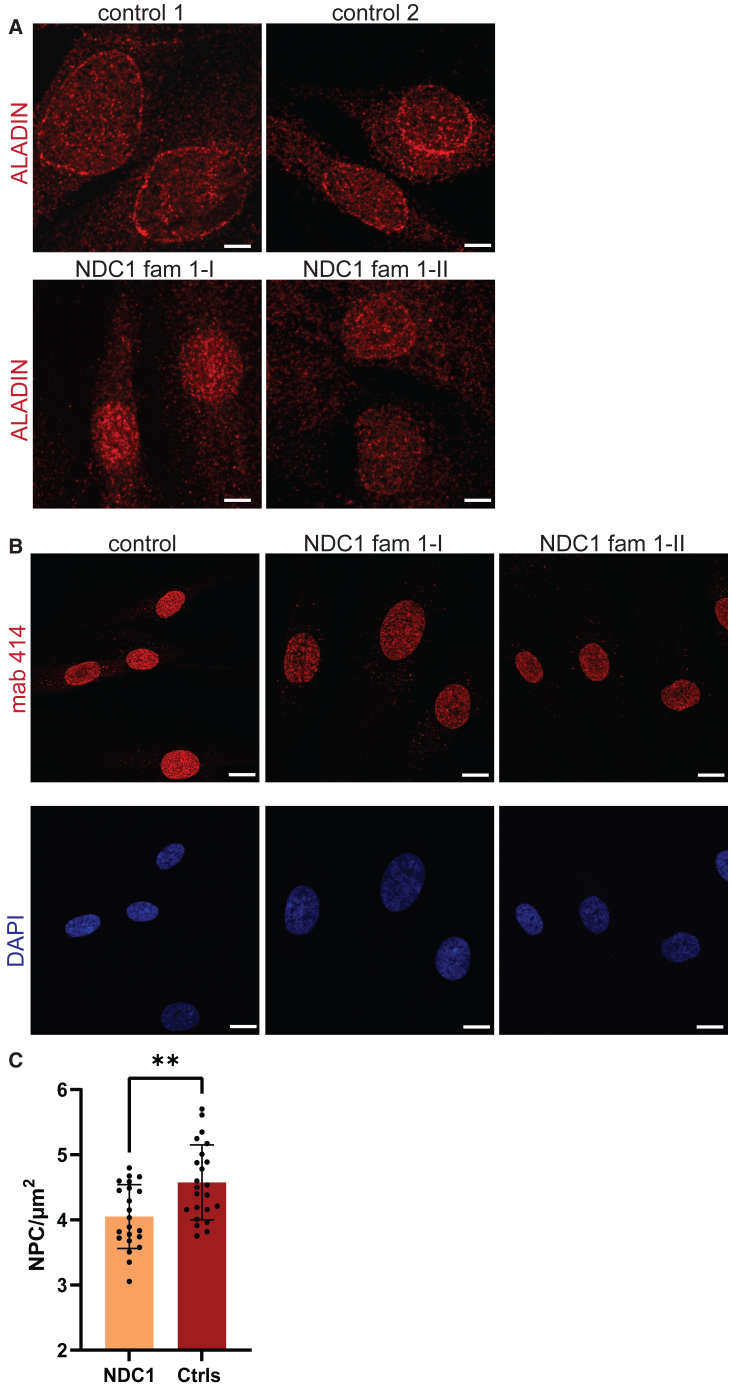
Nuclear pore complexes (NPCs) regulate nucleocytoplasmic transport and are anchored in the nuclear envelope by the transmembrane nucleoporin NDC1. NDC1 is essential for post-mitotic NPC assembly and the recruitment of ALADIN to the nuclear envelope. While no human disorder has been associated to one of the three transmembrane nucleoporins, biallelic variants in AAAS, encoding ALADIN, cause triple A syndrome (Allgrove syndrome). Triple A syndrome, characterized by alacrima, achalasia, and adrenal insufficiency, often includes progressive demyelinating polyneuropathy and other neurological complaints. In this report, diagnostic exome and/or RNA sequencing was performed in seven individuals from four unrelated consanguineous families with AAAS-negative triple A syndrome. Molecular and clinical studies followed to elucidate the pathogenic mechanism. The affected individuals presented with intellectual disability, motor impairment, severe demyelinating with secondary axonal polyneuropathy, alacrima, and achalasia. None of the affected individuals has adrenal insufficiency. All individuals presented with biallelic NDC1 in-frame deletions or missense variants that affect amino acids and protein domains required for ALADIN binding. No other significant variants associated with the phenotypic features were reported. Skin fibroblasts derived from affected individuals show decreased recruitment of ALADIN to the NE and decreased post-mitotic NPC insertion, confirming pathogenicity of the variants. Taken together, our results implicate biallelic NDC1 variants in the pathogenesis of polyneuropathy and a triple A-like disorder without adrenal insufficiency, by interfering with physiological NDC1 functions, including the recruitment of ALADIN to the NPC.
Keywords: ALADIN; NDC1; achalasia; alacrima; neurodevelopmental disorder; nuclear pore complex; peripheral neuropathy; triple A syndrome.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Muir A.M., Cohen J.L., Sheppard S.E., Guttipatti P., Lo T.Y., Weed N., Doherty D., DeMarzo D., Fagerberg C.R., Kjærsgaard L., et al. Bi-allelic Loss-of-Function Variants in NUP188 Cause a Recognizable Syndrome Characterized by Neurologic, Ocular, and Cardiac Abnormalities. Am. J. Hum. Genet. 2020;106:623–631. doi: 10.1016/j.ajhg.2020.03.009. - DOI - PMC - PubMed
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
