

This is a preprint.
Analysis of RNA translation with a deep learning architecture provides new insight into translation control
- PMID: 39005319
- PMCID: PMC11244891
- DOI: 10.1101/2023.07.08.548206
Analysis of RNA translation with a deep learning architecture provides new insight into translation control
Update in
-
Analysis of RNA translation with a deep learning architecture provides new insight into translation control.Nucleic Acids Res. 2025 Apr 10;53(7):gkaf277. doi: 10.1093/nar/gkaf277. Nucleic Acids Res. 2025. PMID: 40219965 Free PMC article.
Abstract
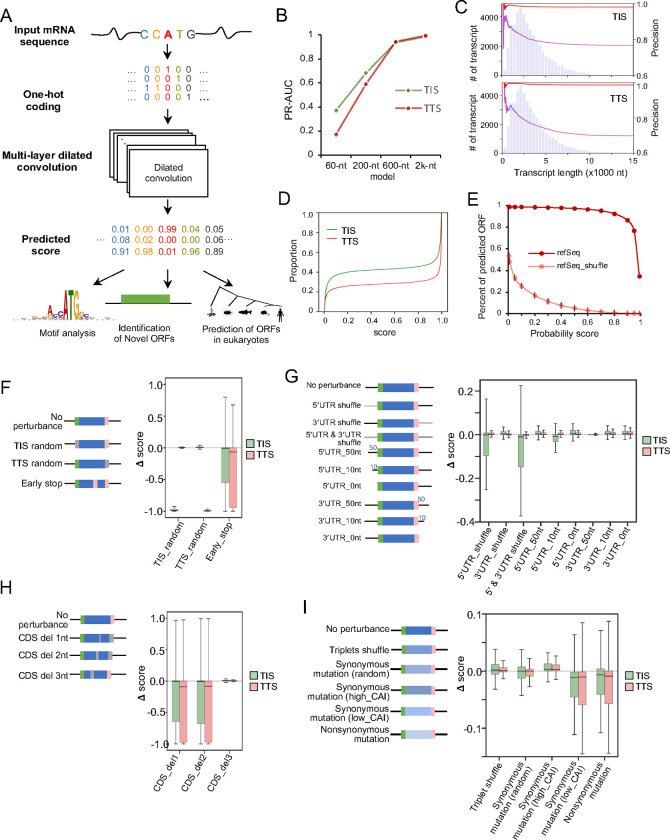
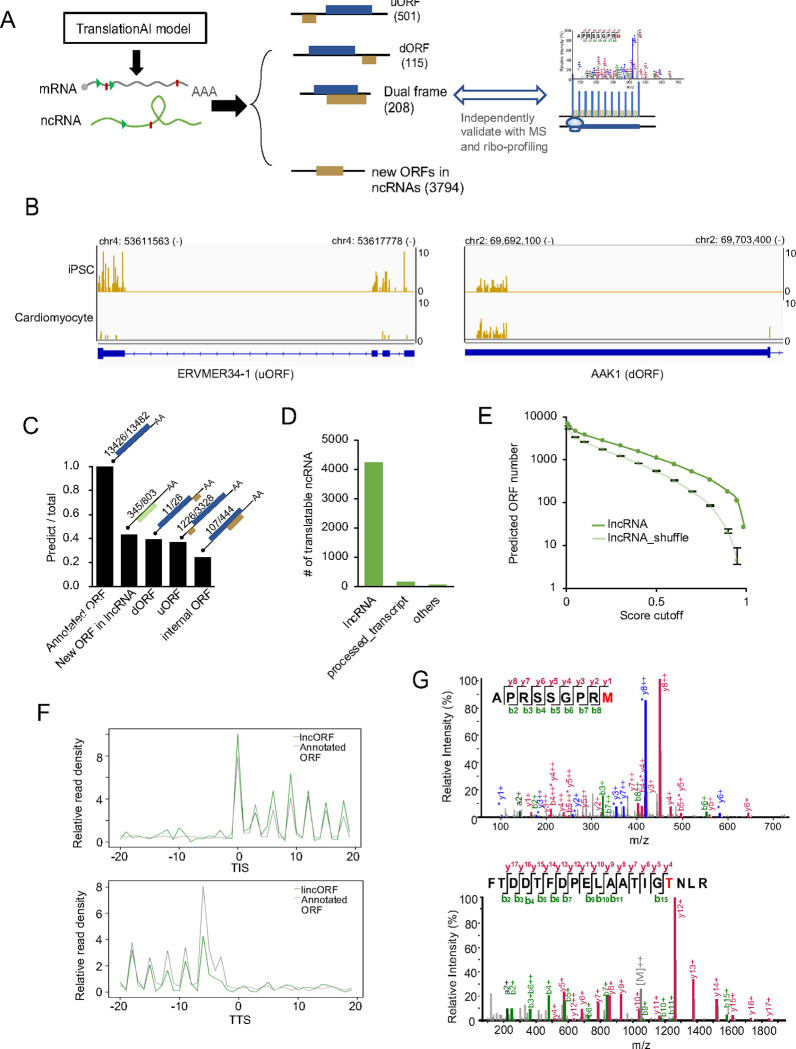
Accurate annotation of coding regions in RNAs is essential for understanding gene translation. We developed a deep neural network to directly predict and analyze translation initiation and termination sites from RNA sequences. Trained with human transcripts, our model learned hidden rules of translation control and achieved a near perfect prediction of canonical translation sites across entire human transcriptome. Surprisingly, this model revealed a new role of codon usage in regulating translation termination, which was experimentally validated. We also identified thousands of new open reading frames in mRNAs or lncRNAs, some of which were confirmed experimentally. The model trained with human mRNAs achieved high prediction accuracy of canonical translation sites in all eukaryotes and good prediction in polycistronic transcripts from prokaryotes or RNA viruses, suggesting a high degree of conservation in translation control. Collectively, we present a general and efficient deep learning model for RNA translation, generating new insights into the complexity of translation regulation.
Conflict of interest statement
Declaration of Interests The authors declare no competing financial interests.
Figures


References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources