

This is a preprint.
A side-by-side comparison of variant function measurements using deep mutational scanning and base editing
- PMID: 39005366
- PMCID: PMC11244880
- DOI: 10.1101/2024.06.30.601444
A side-by-side comparison of variant function measurements using deep mutational scanning and base editing
Update in
-
A side-by-side comparison of variant function measurements using deep mutational scanning and base editing.Nucleic Acids Res. 2025 Jul 19;53(14):gkaf738. doi: 10.1093/nar/gkaf738. Nucleic Acids Res. 2025. PMID: 40744495 Free PMC article.
Abstract
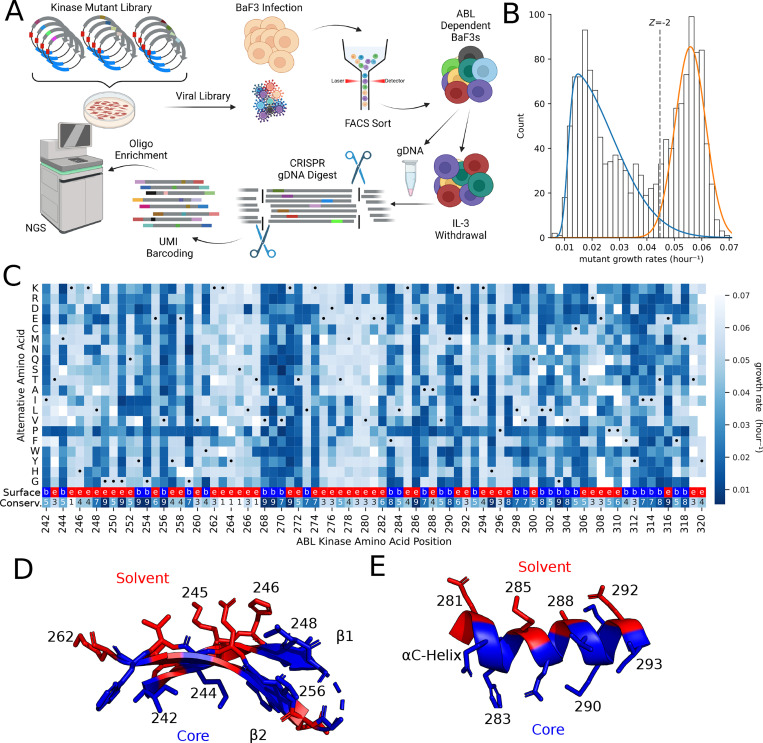
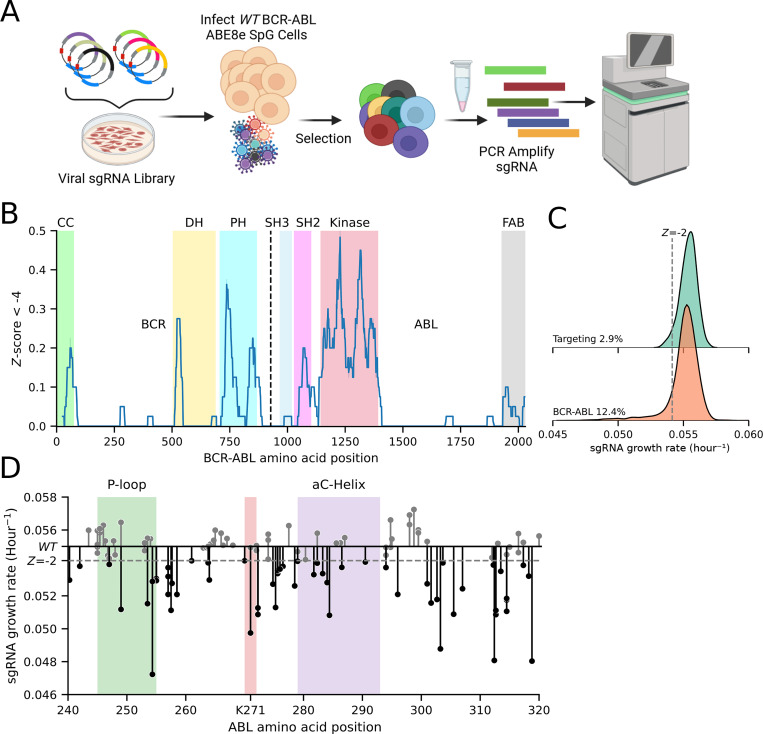
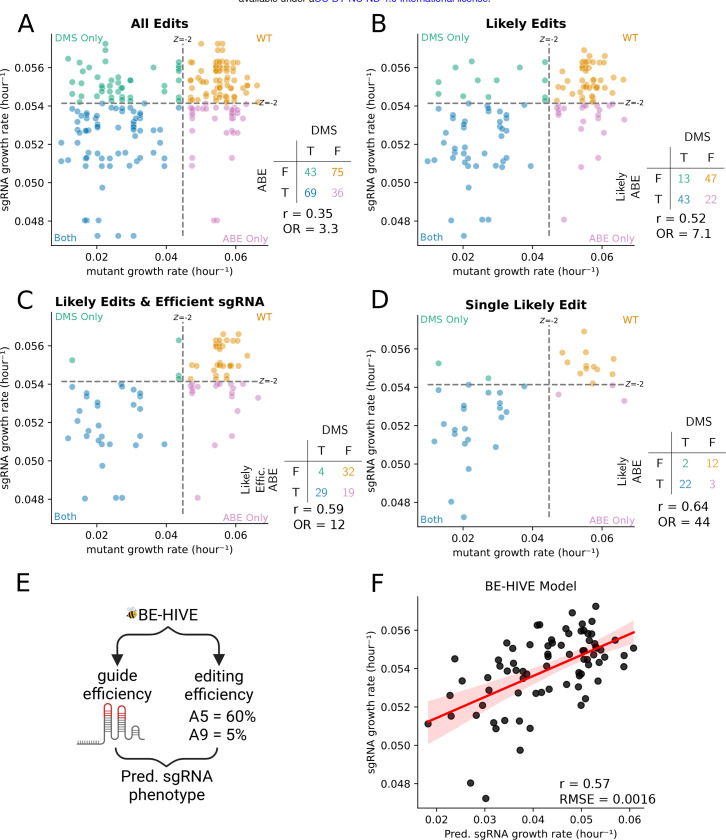
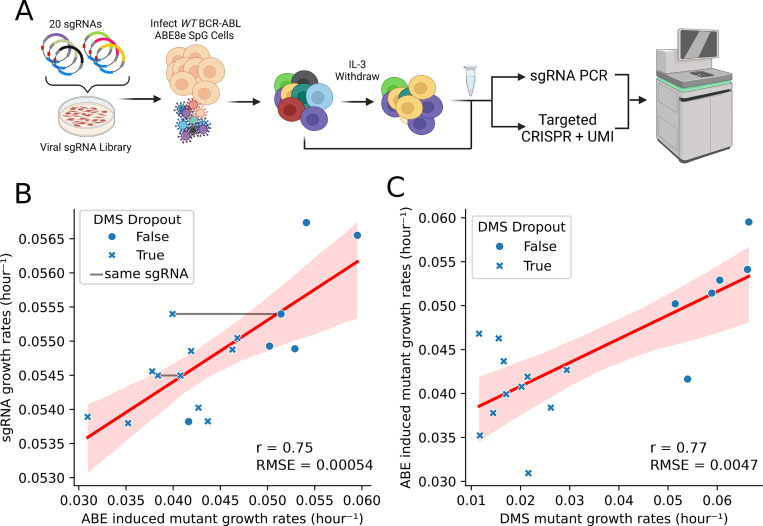
Variant annotation is a crucial objective in mammalian functional genomics. Deep Mutational Scanning (DMS) is a well-established method for annotating human gene variants, but CRISPR base editing (BE) is emerging as an alternative. However, questions remain about how well high-throughput base editing measurements can annotate variant function and the extent of downstream experimental validation required. This study presents the first direct comparison of DMS and BE in the same lab and cell line. Results indicate that focusing on the most likely edits and highest efficiency sgRNAs enhances the agreement between a "gold standard" DMS dataset and a BE screen. A simple filter for sgRNAs making single edits in their window could sufficiently annotate a large proportion of variants directly from sgRNA sequencing of large pools. When multi-edit guides are unavoidable, directly measuring the variants created in the pool, rather than sgRNA abundance, can recover high-quality variant annotation measurements in multiplexed pools. Taken together, our data show a surprising degree of correlation between base editor data and gold standard deep mutational scanning.
Conflict of interest statement
JRP is a co-founder of RedAce Bio. JRP was a co-founder and consultant for Theseus Pharmaceuticals. JRP held equity in Theseus Pharmaceuticals. JRP holds equity in MOMA therapeutics and RedAce Bio. JRP has consulted/consults for MOMA therapeutics, Curie.Bio, Third Rock Ventures, Takeda Pharmaceuticals, Galapagos Pharmaceuticals, and Roche/Genentech. JRP has received honoraria and travel expenses from Roche/Genentech, Third Rock Ventures, and Theseus Pharmaceuticals.
Figures
References
-
- Kinney J.B. and McCandlish D.M. (2019) Massively Parallel Assays and Quantitative Sequence-Function Relationships. Annu Rev Genomics Hum Genet, 20, 99–127. - PubMed
-
- Elbashir S.M., Harborth J., Lendeckel W., Yalcin A., Weber K. and Tuschl T. (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 411, 494–498. - PubMed
-
- Root D.E., Hacohen N., Hahn W.C., Lander E.S. and Sabatini D.M. (2006) Genome-scale loss-of-function screening with a lentiviral RNAi library. Nat Methods, 3, 715–719. - PubMed
-
- Fire A., Xu S., Montgomery M.K., Kostas S.A., Driver S.E. and Mello C.C. (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature, 391, 806–811. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous