

Case Report: Common variable immunodeficiency phenotype and granulomatous-lymphocytic interstitial lung disease with a novel SOCS1 variant
- PMID: 39005503
- PMCID: PMC11239428
- DOI: 10.3389/fped.2024.1423858
Case Report: Common variable immunodeficiency phenotype and granulomatous-lymphocytic interstitial lung disease with a novel SOCS1 variant
Abstract
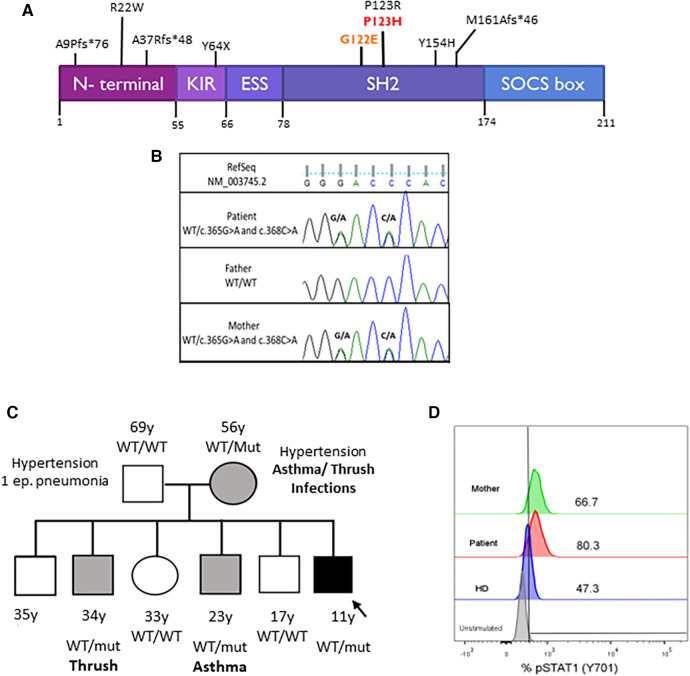
Common variable immunodeficiency is a heterogeneous symptomatic group of inborn errors of immunity that mainly affects antibodies production and/or function, predisposing patients to recurrent and severe infections. More than half of them usually develop autoimmunity, lymphoproliferation, enteropathy, and malignancies. Among these conditions, chronic lung disease such as granulomatous-lymphocytic interstitial lung disease is one of the leading causes of death in these patients. Recently, many genes that play a key role in B and T cells' development, maintenance, and/or cytokines signaling pathways have been implicated in the pathogenesis of the disease. Here, we describe the first Argentinian patient presenting with common variable immunodeficiency and granulomatous-lymphocytic interstitial lung disease, harboring two in cis heterozygous variants in the SOCS1 gene.
Keywords: GLILD; SOCS1; common variable immunodeficiency; granulomatous–lymphocytic interstitial lung disease; inborn errors of immunity.
© 2024 Caldirola, Daiana, Gomez Raccio, García, Bernacchia, Medín, Gaillard and Di Giovanni.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
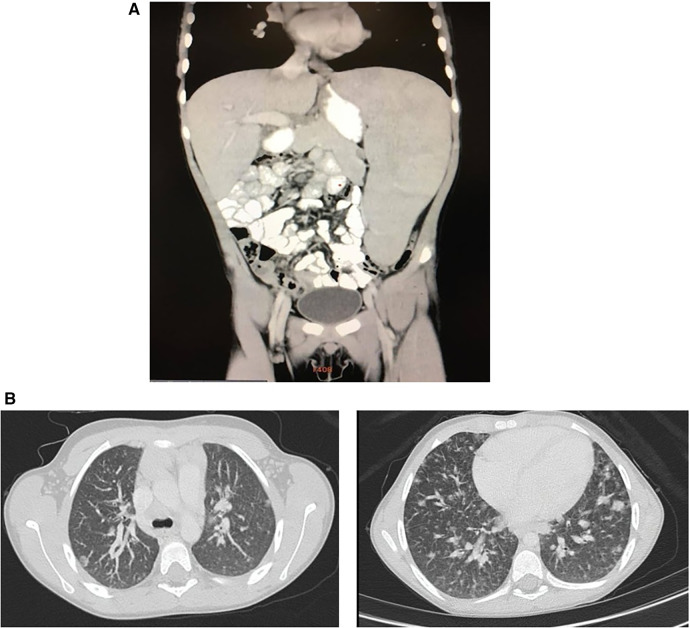
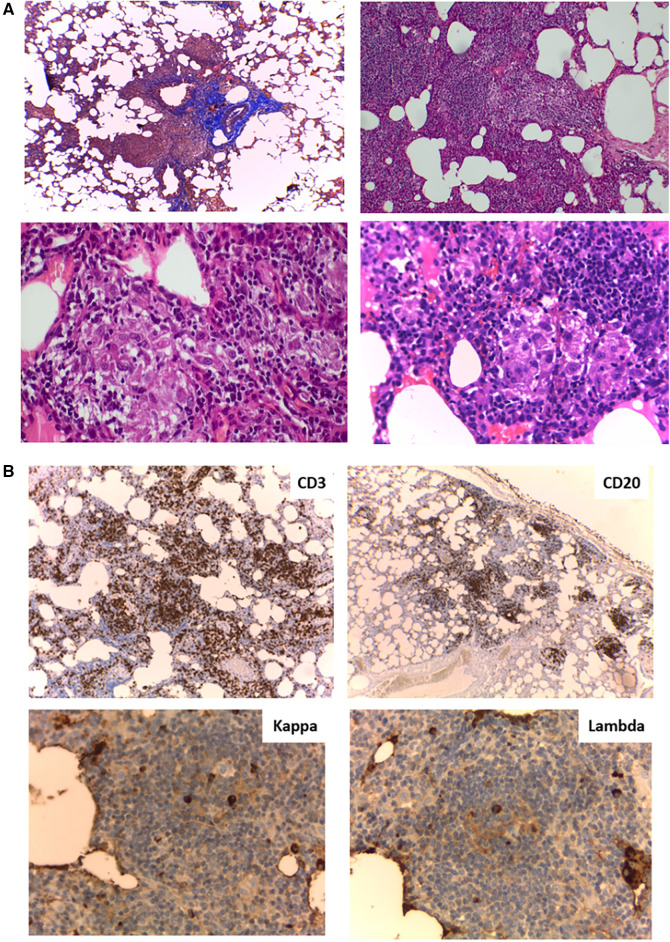
Figures
References
-
- Sullivan K. Stiehm’s Immune Deficiencies. Waltham, MA: Elsevier; (2020).
Publication types
LinkOut - more resources
Full Text Sources