

Clinical presentation, diagnosis, and treatment of chronic granulomatous disease
- PMID: 39005504
- PMCID: PMC11239527
- DOI: 10.3389/fped.2024.1384550
Clinical presentation, diagnosis, and treatment of chronic granulomatous disease
Abstract
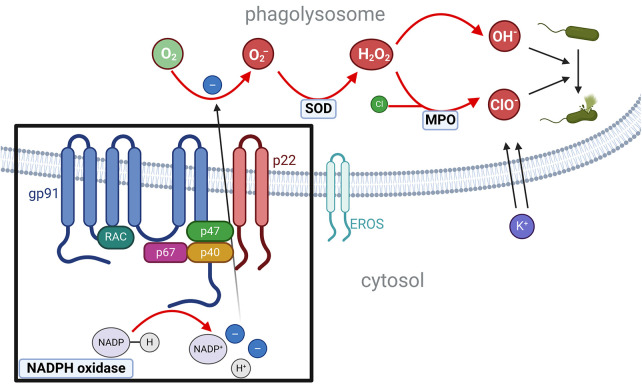
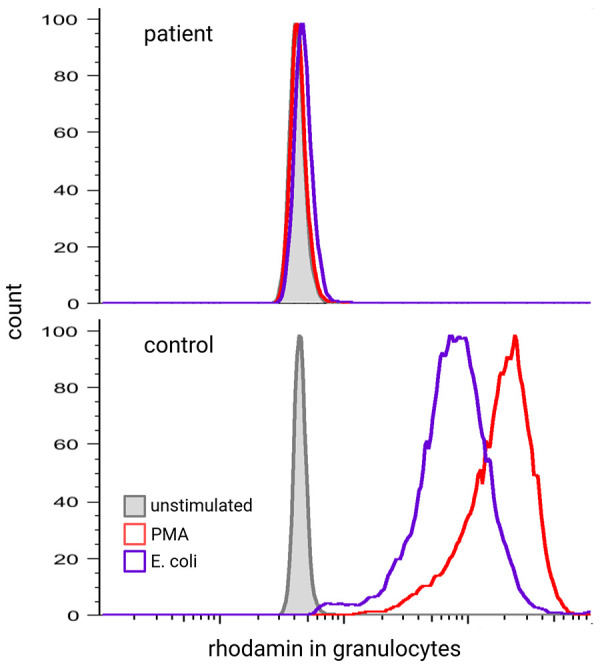
Chronic granulomatous disease (CGD) is caused by an impaired respiratory burst reaction in phagocytes. CGD is an X-linked (XL) (caused by pathogenic variants in CYBB) or autosomal recessive inborn error of immunity (caused by pathogenic variants in CYBA, NCF1, NCF2, or CYBC1). Female carriers of XL-CGD and unfavorable lyonization may present with the partial or full picture of CGD. Patients with CGD are at increased risk for invasive bacterial and fungal infections of potentially any organ, but especially the lymph nodes, liver, and lungs. Pathogens most frequently isolated are S. aureus and Aspergillus spp. Autoinflammation is difficult to control with immunosuppression, and patients frequently remain dependent on steroids. To diagnose CGD, reactive oxygen intermediates (O2 - or H2O2) generated by the NADPH oxidase in peripheral blood phagocytes are measured upon in vitro activation with either phorbol-12-myristate-13-acetate (PMA) and/or TLR4 ligands (E. coli or LPS). Conservative treatment requires strict hygienic conduct and adherence to antibiotic prophylaxis against bacteria and fungi, comprising cotrimoxazole and triazoles. The prognosis of patients treated conservatively is impaired: for the majority of patients, recurrent and/or persistent infections, autoinflammation, and failure to thrive remain lifelong challenges. In contrast, cellular therapies (allogeneic stem cell transplantation or gene therapy) can cure CGD. Optimal outcomes in cellular therapies are observed in individuals without ongoing infections or inflammation. Yet cellular therapies are the only curative option for patients with persistent fungal infections or autoinflammation.
Keywords: HSCT; chronic granolumatous disease; clinical presentation; diagnosis; hematopoietic stem cell transplantation; therapy.
© 2024 Staudacher and von Bernuth.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures


Similar articles
-
Clinical, functional, and genetic characterization of chronic granulomatous disease in 89 Turkish patients.J Allergy Clin Immunol. 2013 Nov;132(5):1156-1163.e5. doi: 10.1016/j.jaci.2013.05.039. Epub 2013 Jul 31. J Allergy Clin Immunol. 2013. PMID: 23910690
-
Chronic granulomatous disease: Clinical, molecular, and therapeutic aspects.Pediatr Allergy Immunol. 2016 May;27(3):242-53. doi: 10.1111/pai.12527. Epub 2016 Jan 21. Pediatr Allergy Immunol. 2016. PMID: 26680691 Review.
-
Chronic Granulomatous Disease: a Comprehensive Review.Clin Rev Allergy Immunol. 2021 Oct;61(2):101-113. doi: 10.1007/s12016-020-08800-x. Clin Rev Allergy Immunol. 2021. PMID: 32524254 Review.
-
Molecular Challenges in the Diagnosis of X-Linked Chronic Granulomatous Disease: CNVs, Intronic Variants, Skewed X-Chromosome Inactivation, and Gonosomal Mosaicism.J Clin Immunol. 2023 Nov;43(8):1953-1963. doi: 10.1007/s10875-023-01556-x. Epub 2023 Aug 19. J Clin Immunol. 2023. PMID: 37597073
-
[Bacillus Calmette-Guérin infection and chronic granulomatous disease due to new pathogenic variants in the NCF2 gene in the Mayan ethnic group. Report of two cases.].Rev Alerg Mex. 2023 Apr 19;69(4):220-227. doi: 10.29262/ram.v69i4.1145. Rev Alerg Mex. 2023. PMID: 37218049 Spanish.
Cited by
-
Diagnosis of Chronic Granulomatous Disease: Strengths and Challenges in the Genomic Era.J Clin Med. 2024 Jul 29;13(15):4435. doi: 10.3390/jcm13154435. J Clin Med. 2024. PMID: 39124702 Free PMC article. Review.
-
Human immunity to fungal infections.J Exp Med. 2025 Jun 2;222(6):e20241215. doi: 10.1084/jem.20241215. Epub 2025 Apr 15. J Exp Med. 2025. PMID: 40232283 Review.
-
Liver Abscess: Think Outside the Box.Cureus. 2025 Jun 25;17(6):e86755. doi: 10.7759/cureus.86755. eCollection 2025 Jun. Cureus. 2025. PMID: 40718299 Free PMC article.
References
-
- Janeway CA, Craig J, Davidson M, Downey W, Gitlin D, Sullivan JC. Hypergammaglobulinemia associated with severe, recurrent and chronic non-specific infection. Am J Dis Child. (1954) 88:388–92.
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous