

Top-down proteomics
- PMID: 39006170
- PMCID: PMC11242913
- DOI: 10.1038/s43586-024-00318-2
Top-down proteomics
Abstract
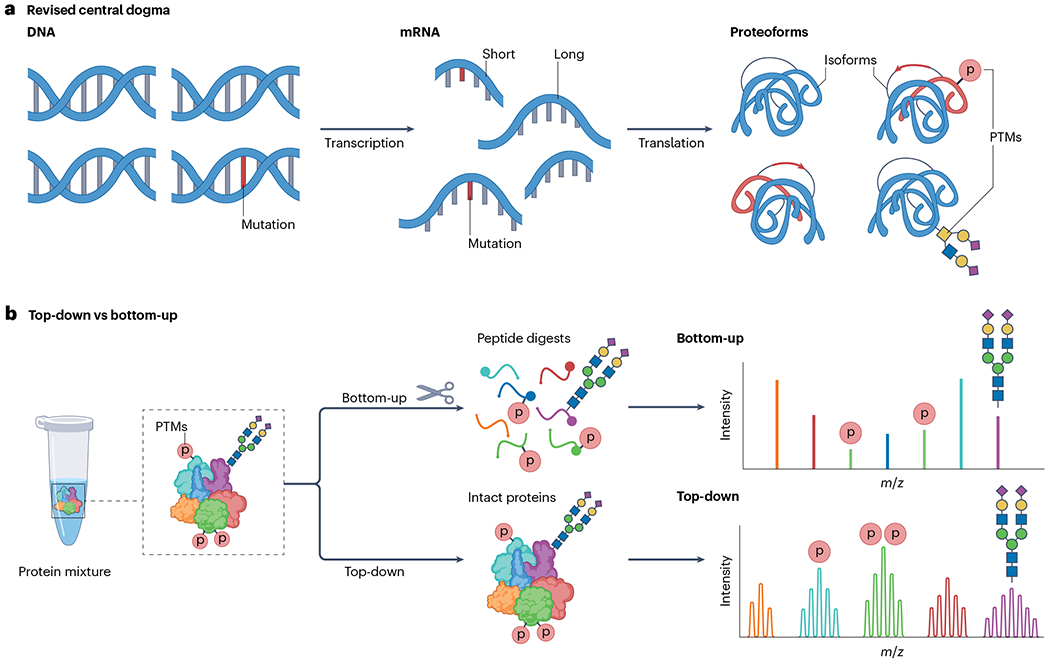
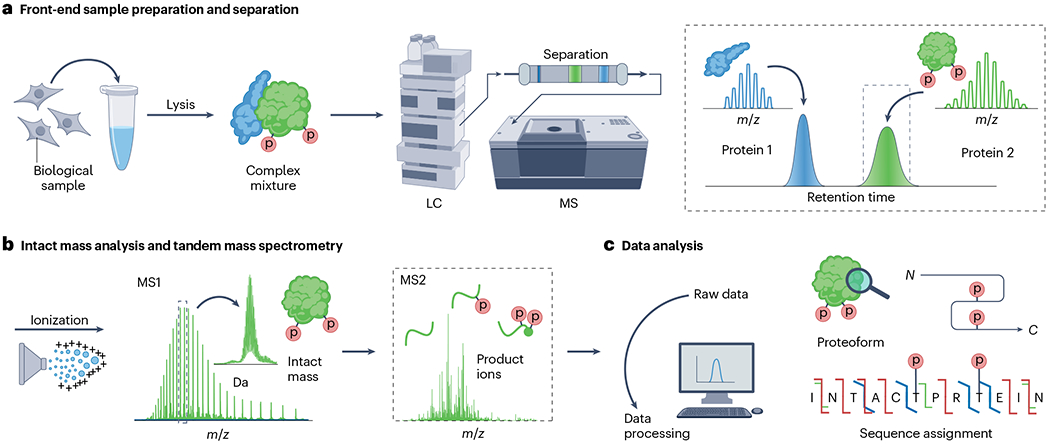
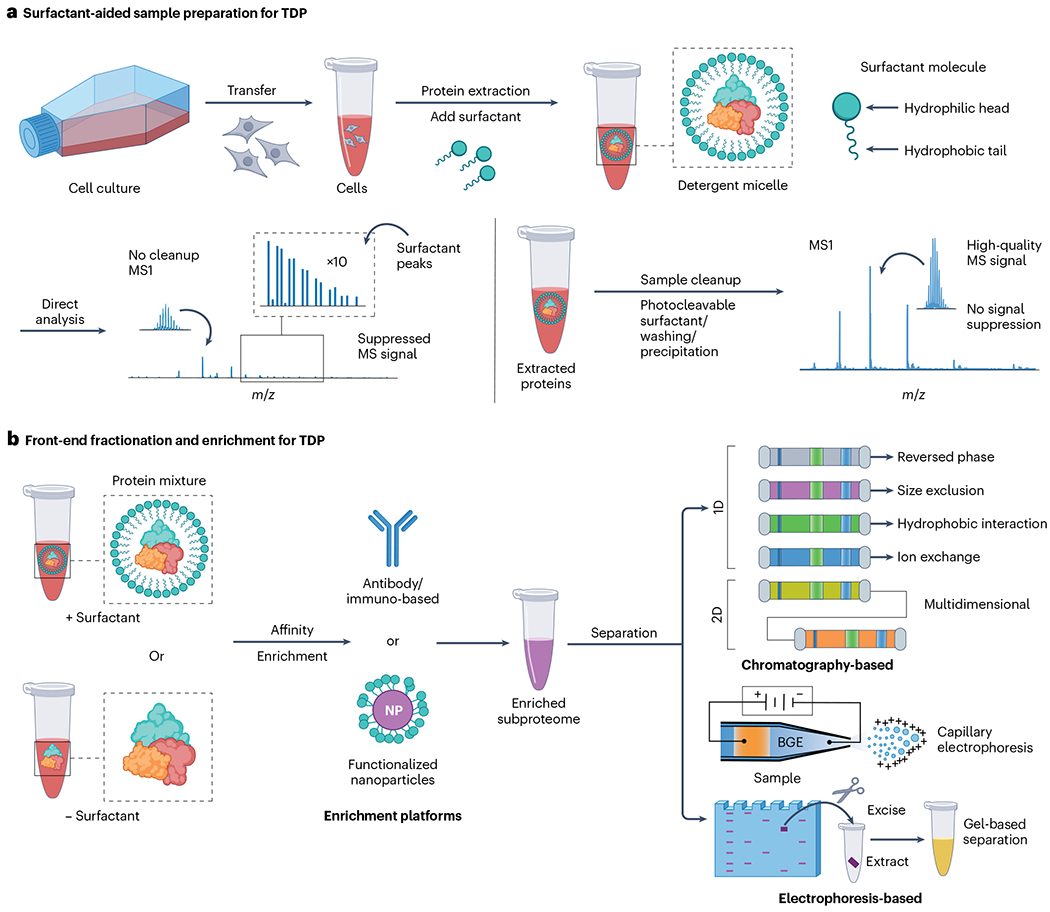
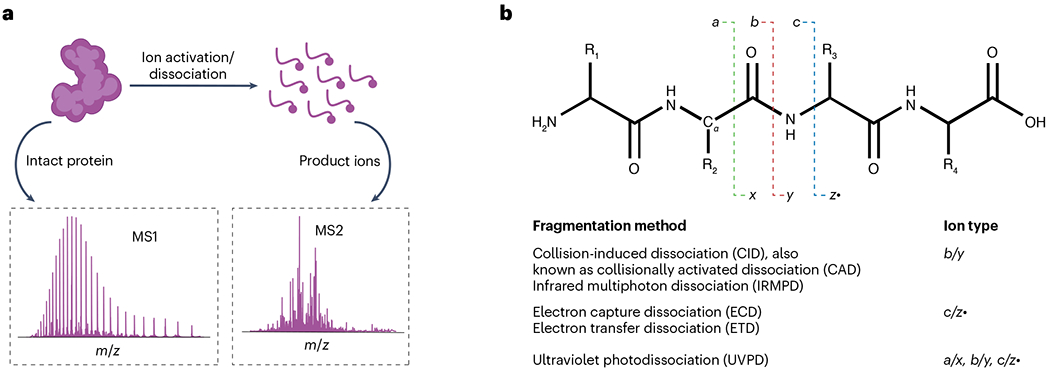
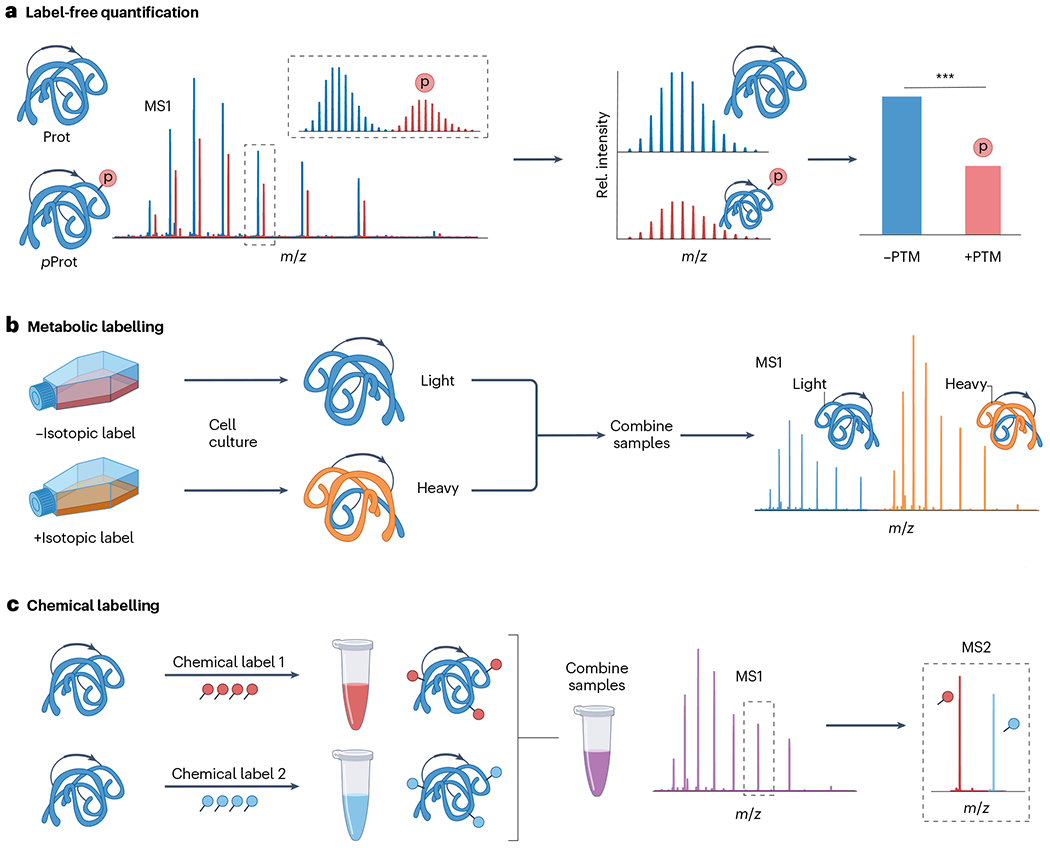
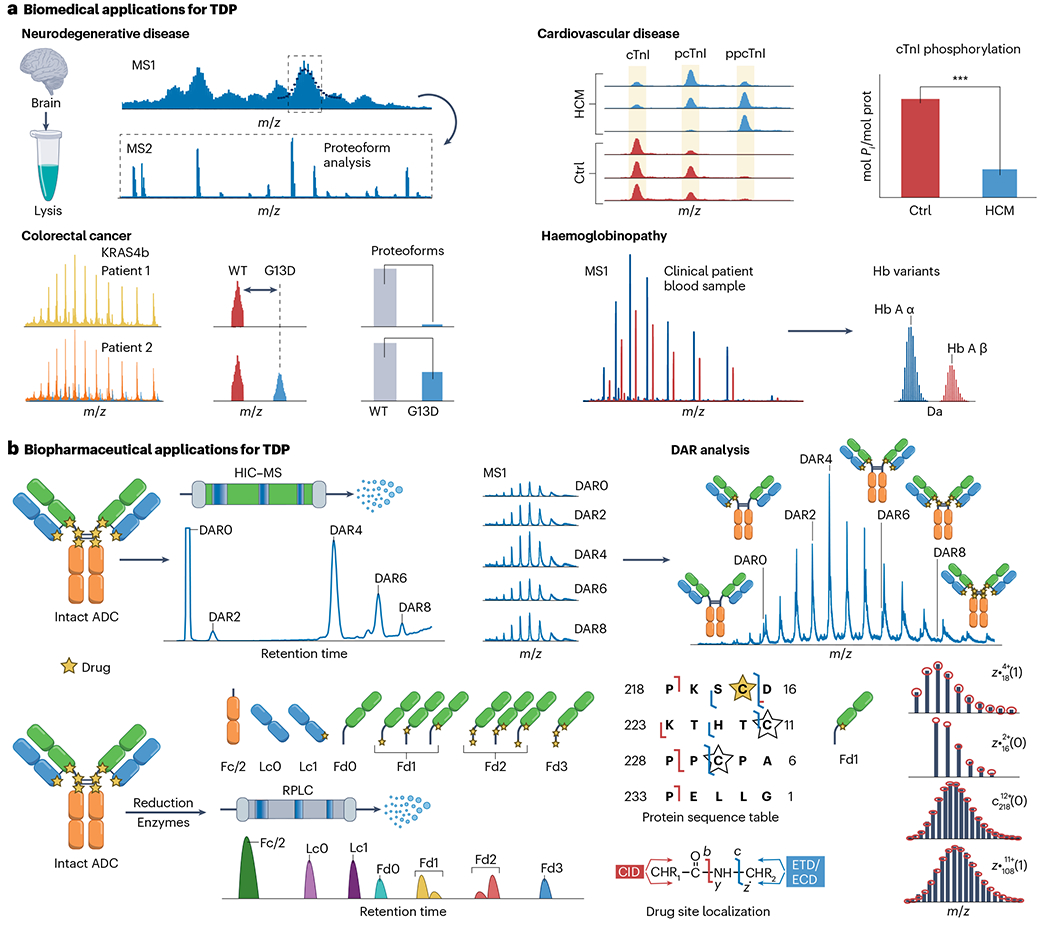
Proteoforms, which arise from post-translational modifications, genetic polymorphisms and RNA splice variants, play a pivotal role as drivers in biology. Understanding proteoforms is essential to unravel the intricacies of biological systems and bridge the gap between genotypes and phenotypes. By analysing whole proteins without digestion, top-down proteomics (TDP) provides a holistic view of the proteome and can decipher protein function, uncover disease mechanisms and advance precision medicine. This Primer explores TDP, including the underlying principles, recent advances and an outlook on the future. The experimental section discusses instrumentation, sample preparation, intact protein separation, tandem mass spectrometry techniques and data collection. The results section looks at how to decipher raw data, visualize intact protein spectra and unravel data analysis. Additionally, proteoform identification, characterization and quantification are summarized, alongside approaches for statistical analysis. Various applications are described, including the human proteoform project and biomedical, biopharmaceutical and clinical sciences. These are complemented by discussions on measurement reproducibility, limitations and a forward-looking perspective that outlines areas where the field can advance, including potential future applications.
Conflict of interest statement
Competing interests J.A.L., J.C.-R., J.N.A., L.P.-T., L.M.S. and Y.G. are currently board members of Consortium for Top-down Proteomics. Y.O.T. is an employee of Spectroswiss, a company that develops data acquisition systems and data processing software for mass spectrometry. X.L. has a project contract with Bioinformatics Solutions Inc., a company that develops data processing software for mass spectrometry. D.S.R. and Y.G. are named as inventors for the patent application US Patent App. 17/786,482. L.P.-T. is named as an inventor for the US Patent App. 17/954,834. Y.G. is named as an inventor for the US Patent App. 18/069,005; US Patent App. 17/978,793; US Patent App. 18/451,614; and US Patent 11,567,085. S.W. declares no competing interests.
Figures

References
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous