

Mechanisms of pulmonary endothelial barrier dysfunction in acute lung injury and acute respiratory distress syndrome
- PMID: 39006829
- PMCID: PMC11242916
- DOI: 10.1016/j.pccm.2024.04.002
Mechanisms of pulmonary endothelial barrier dysfunction in acute lung injury and acute respiratory distress syndrome
Abstract
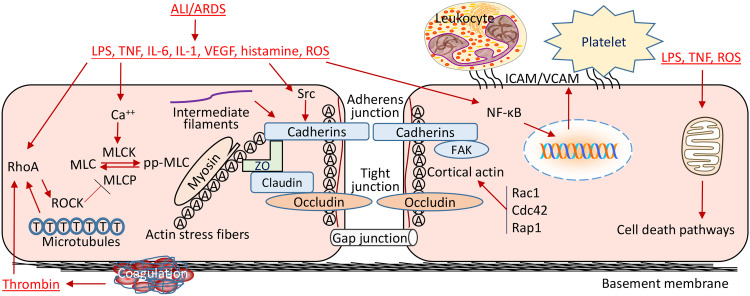
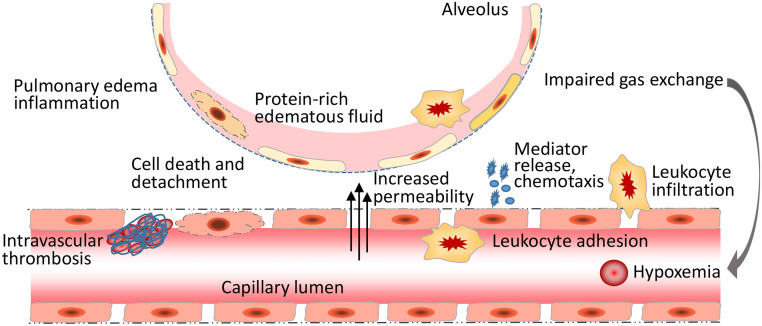
Endothelial cells (ECs) form a semi-permeable barrier between the interior space of blood vessels and the underlying tissues. Pulmonary endothelial barrier integrity is maintained through coordinated cellular processes involving receptors, signaling molecules, junctional complexes, and protein-regulated cytoskeletal reorganization. In acute lung injury (ALI) or its more severe form acute respiratory distress syndrome (ARDS), the loss of endothelial barrier integrity secondary to endothelial dysfunction caused by severe pulmonary inflammation and/or infection leads to pulmonary edema and hypoxemia. Pro-inflammatory agonists such as histamine, thrombin, bradykinin, interleukin 1β, tumor necrosis factor α, vascular endothelial growth factor, angiopoietin-2, and platelet-activating factor, as well as bacterial toxins and reactive oxygen species, cause dynamic changes in cytoskeletal structure, adherens junction disorganization, and detachment of vascular endothelial cadherin (VE-cadherin) from the actin cytoskeleton, leading to an increase in endothelial permeability. Endothelial interactions with leukocytes, platelets, and coagulation enhance the inflammatory response. Moreover, inflammatory infiltration and the associated generation of pro-inflammatory cytokines during infection cause EC death, resulting in further compromise of the structural integrity of lung endothelial barrier. Despite the use of potent antibiotics and aggressive intensive care support, the mortality of ALI is still high, because the mechanisms of pulmonary EC barrier disruption are not fully understood. In this review, we summarized recent advances in the studies of endothelial cytoskeletal reorganization, inter-endothelial junctions, endothelial inflammation, EC death, and endothelial repair in ALI and ARDS, intending to shed some light on the potential diagnostic and therapeutic targets in the clinical management of the disease.
Keywords: Acute lung injury; Acute respiratory distress syndrome; Endothelium; Lung; Pulmonary edema.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
