

Gut microbial factors predict disease severity in a mouse model of multiple sclerosis
- PMID: 39009690
- PMCID: PMC11371644
- DOI: 10.1038/s41564-024-01761-3
Gut microbial factors predict disease severity in a mouse model of multiple sclerosis
Abstract
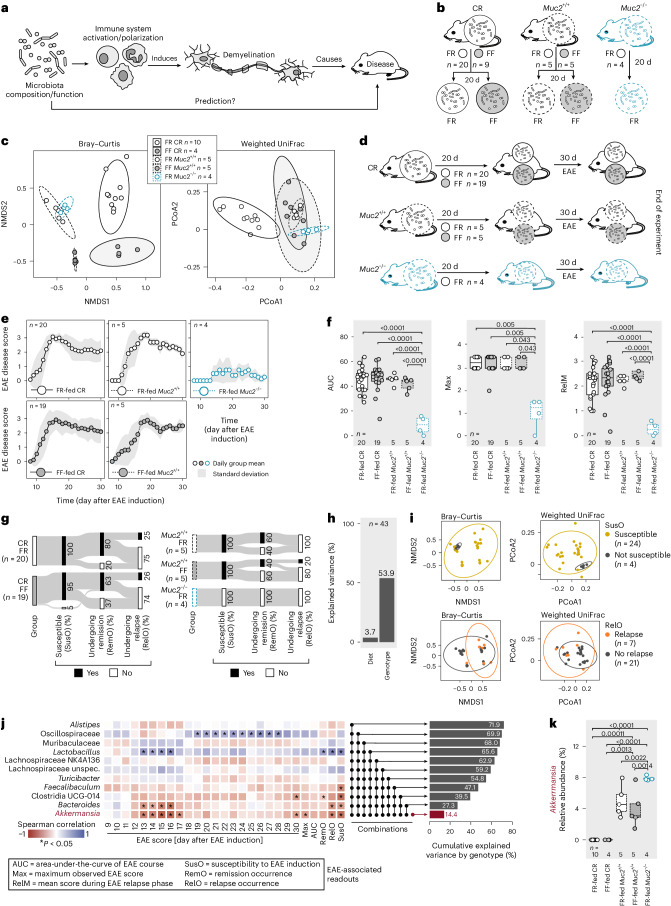
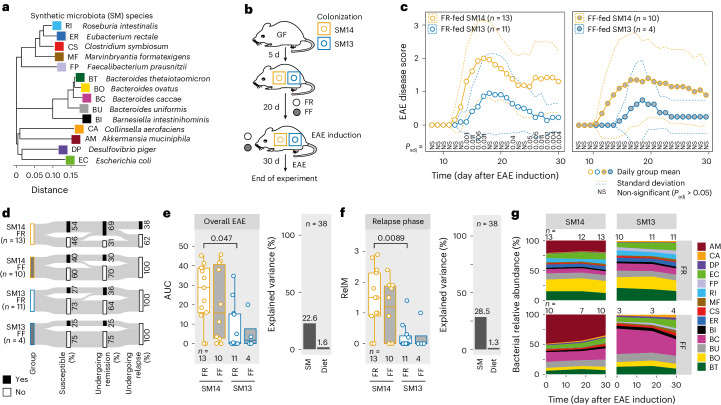
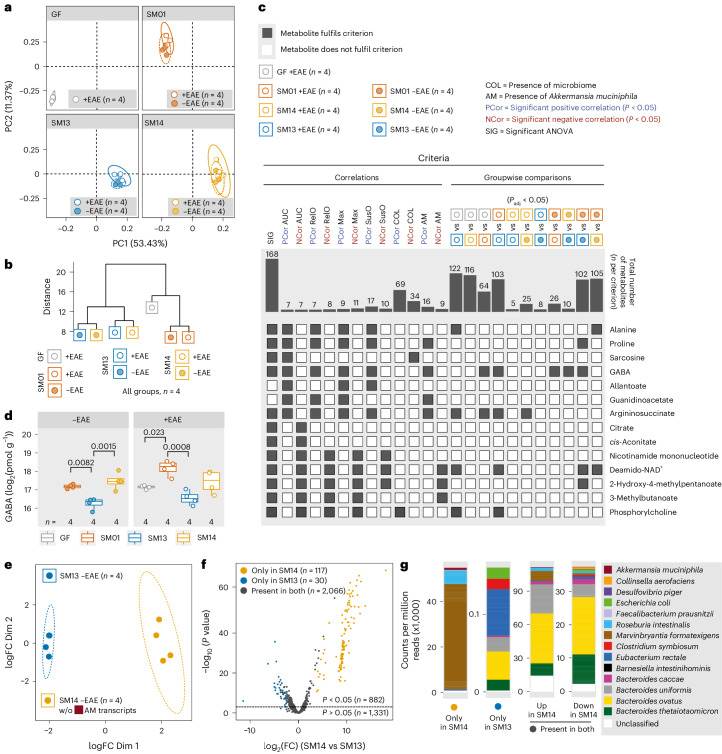
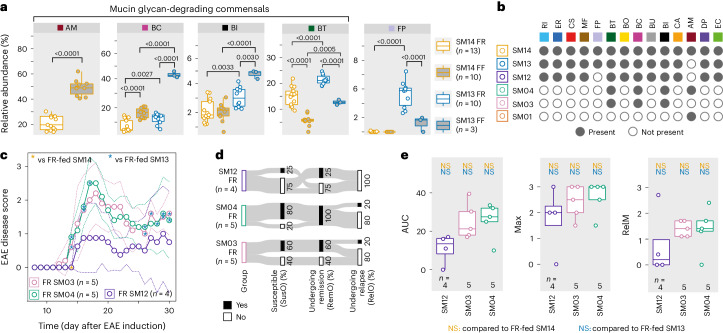
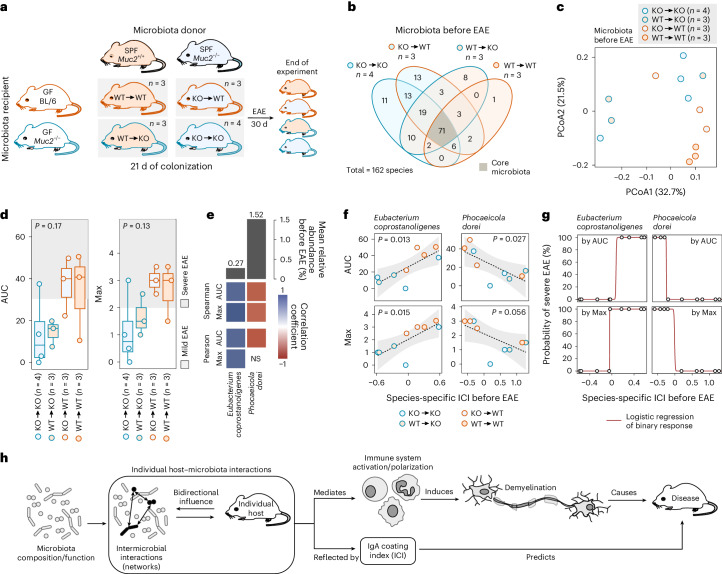
Gut bacteria are linked to neurodegenerative diseases but the risk factors beyond microbiota composition are limited. Here we used a pre-clinical model of multiple sclerosis (MS), experimental autoimmune encephalomyelitis (EAE), to identify microbial risk factors. Mice with different genotypes and complex microbiotas or six combinations of a synthetic human microbiota were analysed, resulting in varying probabilities of severe neuroinflammation. However, the presence or relative abundances of suspected microbial risk factors failed to predict disease severity. Akkermansia muciniphila, often associated with MS, exhibited variable associations with EAE severity depending on the background microbiota. Significant inter-individual disease course variations were observed among mice harbouring the same microbiota. Evaluation of microbial functional characteristics and host immune responses demonstrated that the immunoglobulin A coating index of certain bacteria before disease onset is a robust individualized predictor of disease development. Our study highlights the need to consider microbial community networks and host-specific bidirectional interactions when aiming to predict severity of neuroinflammation.
© 2024. The Author(s).
Conflict of interest statement
M.S.D. works as a consultant and an advisory board member at Theralution GmbH, Germany. S.F. is a founder and CEO of Metagen, Inc., Japan, focused on the design and control of the gut environment for human health. The other authors declare no competing interests.
Figures











Similar articles
-
Pomegranate peel extract ameliorates the severity of experimental autoimmune encephalomyelitis via modulation of gut microbiota.Gut Microbes. 2020 Nov 9;12(1):1857515. doi: 10.1080/19490976.2020.1857515. Gut Microbes. 2020. PMID: 33382357 Free PMC article.
-
Interactions between host genetics and gut microbiota determine susceptibility to CNS autoimmunity.Proc Natl Acad Sci U S A. 2020 Nov 3;117(44):27516-27527. doi: 10.1073/pnas.2002817117. Epub 2020 Oct 19. Proc Natl Acad Sci U S A. 2020. PMID: 33077601 Free PMC article.
-
Mining the microbiota to identify gut commensals modulating neuroinflammation in a mouse model of multiple sclerosis.Microbiome. 2022 Oct 17;10(1):174. doi: 10.1186/s40168-022-01364-2. Microbiome. 2022. PMID: 36253847 Free PMC article.
-
Gut microbiota in multiple sclerosis and animal models.FEBS J. 2025 Mar;292(6):1330-1356. doi: 10.1111/febs.17161. Epub 2024 May 30. FEBS J. 2025. PMID: 38817090 Review.
-
Gut Microbiota in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis: Current Applications and Future Perspectives.Mediators Inflamm. 2018 Apr 2;2018:8168717. doi: 10.1155/2018/8168717. eCollection 2018. Mediators Inflamm. 2018. PMID: 29805314 Free PMC article. Review.
Cited by
-
Akkermansia mono-colonization modulates microglia and astrocytes in a strain specific manner.J Neuroinflammation. 2025 Mar 27;22(1):94. doi: 10.1186/s12974-025-03417-3. J Neuroinflammation. 2025. PMID: 40148962 Free PMC article.
-
Human Untargeted Metabolomics in High-Throughput Gut Microbiome Research: Ethanol vs Methanol.Anal Chem. 2025 Mar 11;97(9):4945-4953. doi: 10.1021/acs.analchem.4c05142. Epub 2025 Feb 27. Anal Chem. 2025. PMID: 40015251 Free PMC article.
-
Lactobacillus helveticus mitigates diarrhea and inflammation induced by enterotoxigenic E. coli through rebalance of gut microbiota.Curr Res Food Sci. 2025 Jul 17;11:101147. doi: 10.1016/j.crfs.2025.101147. eCollection 2025. Curr Res Food Sci. 2025. PMID: 40735631 Free PMC article.
-
The unresolved struggle of 16S rRNA amplicon sequencing: a benchmarking analysis of clustering and denoising methods.Environ Microbiome. 2025 May 13;20(1):51. doi: 10.1186/s40793-025-00705-6. Environ Microbiome. 2025. PMID: 40361240 Free PMC article.
-
Gut microbiota and multiple sclerosis: a potential diagnostic and prognostic marker?Neurodegener Dis Manag. 2024;14(6):189-192. doi: 10.1080/17582024.2024.2435249. Epub 2024 Nov 27. Neurodegener Dis Manag. 2024. PMID: 39600266 No abstract available.
References
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
