

Building a translational cancer dependency map for The Cancer Genome Atlas
- PMID: 39009815
- PMCID: PMC11358024
- DOI: 10.1038/s43018-024-00789-y
Building a translational cancer dependency map for The Cancer Genome Atlas
Abstract
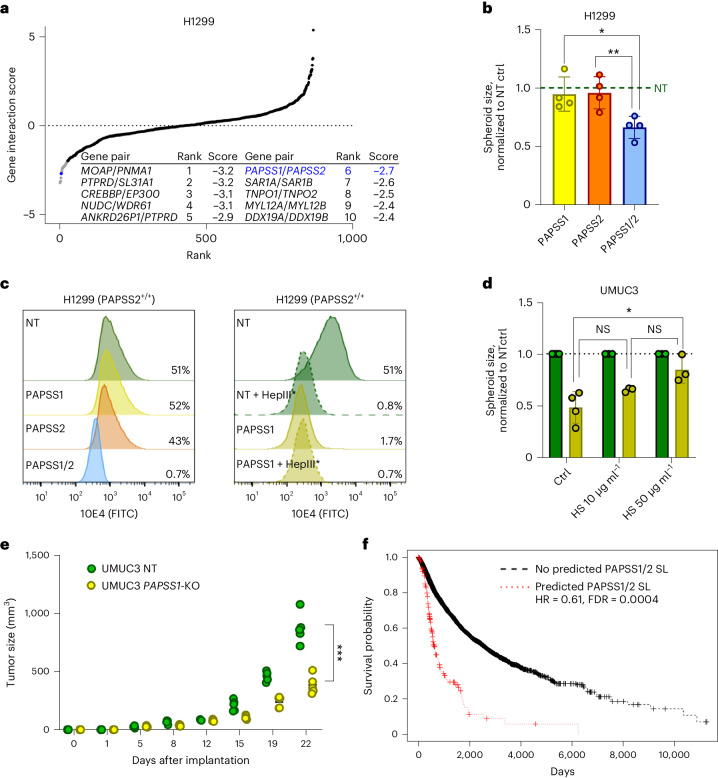
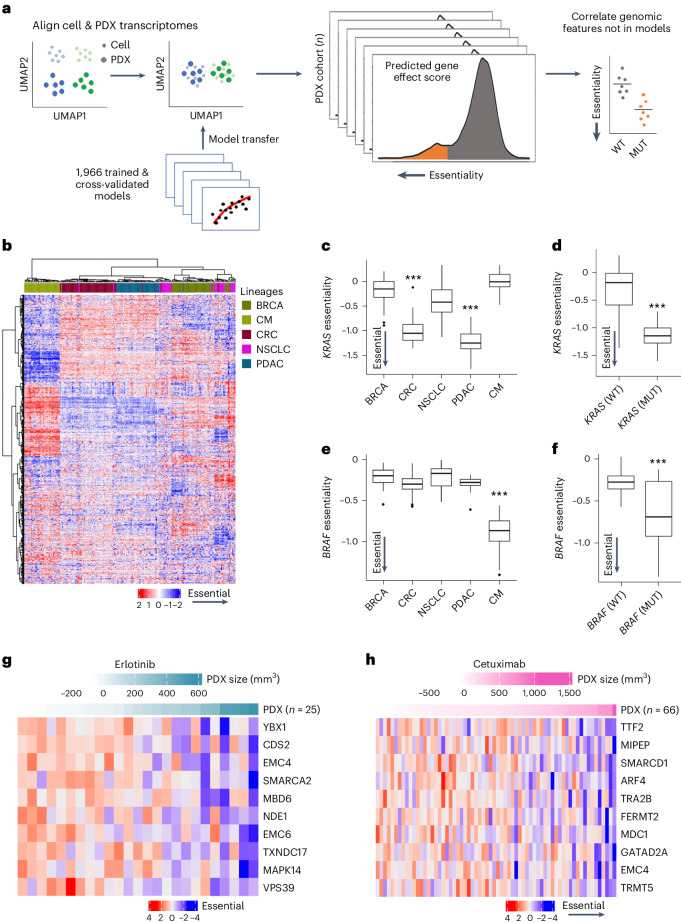
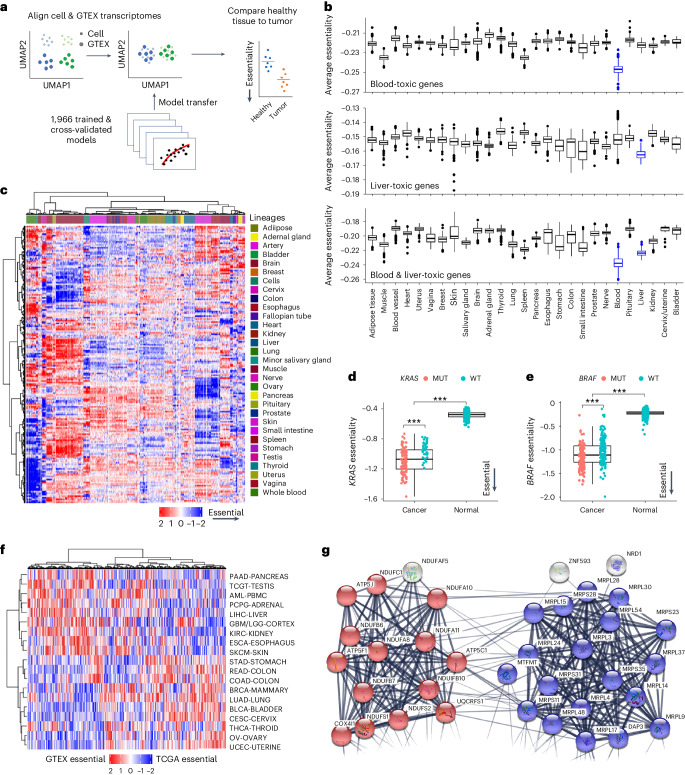
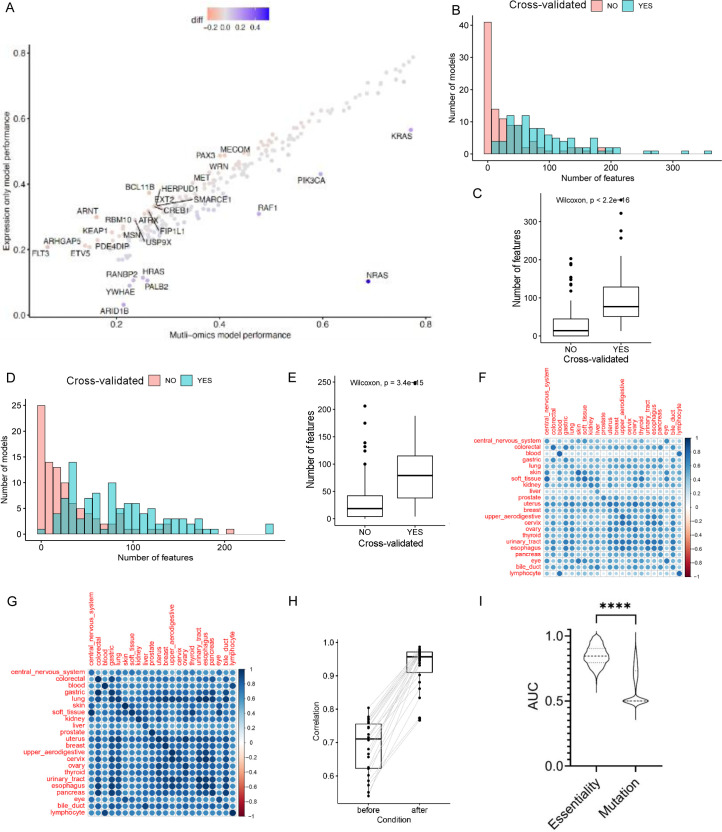
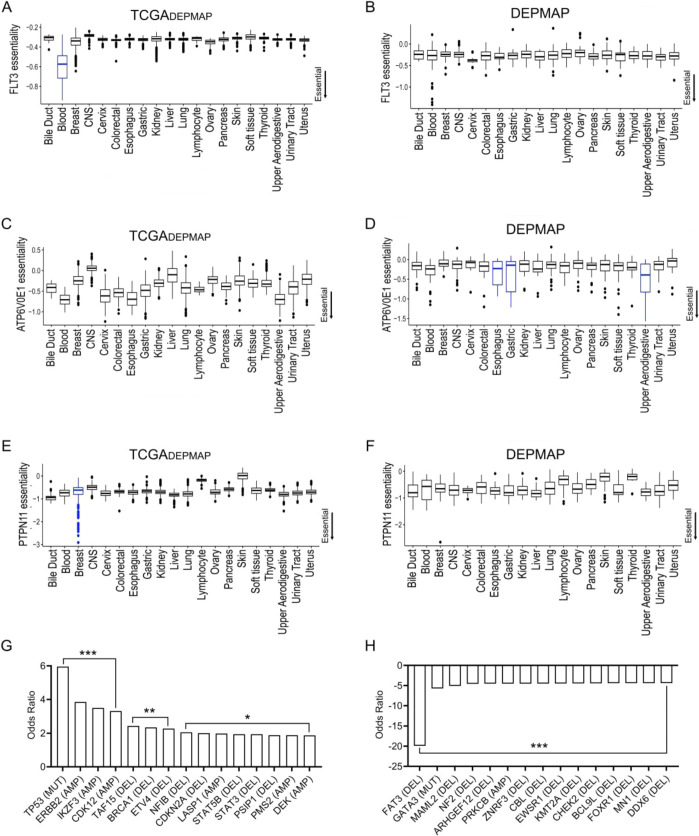
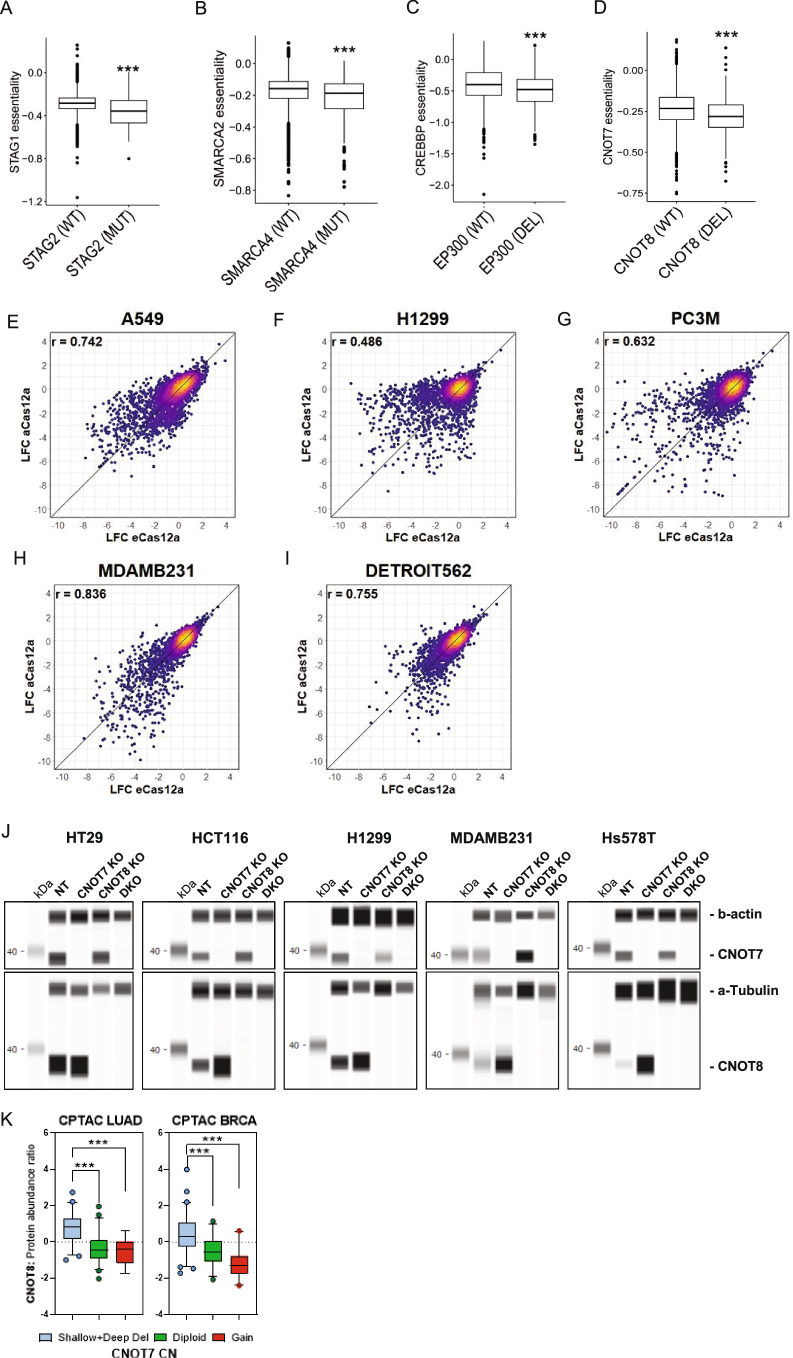
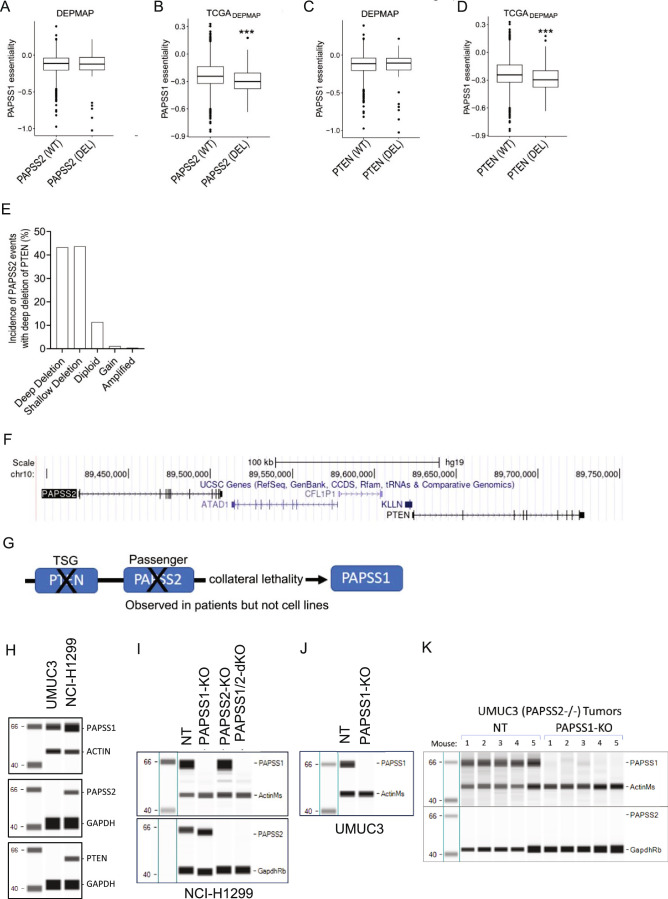
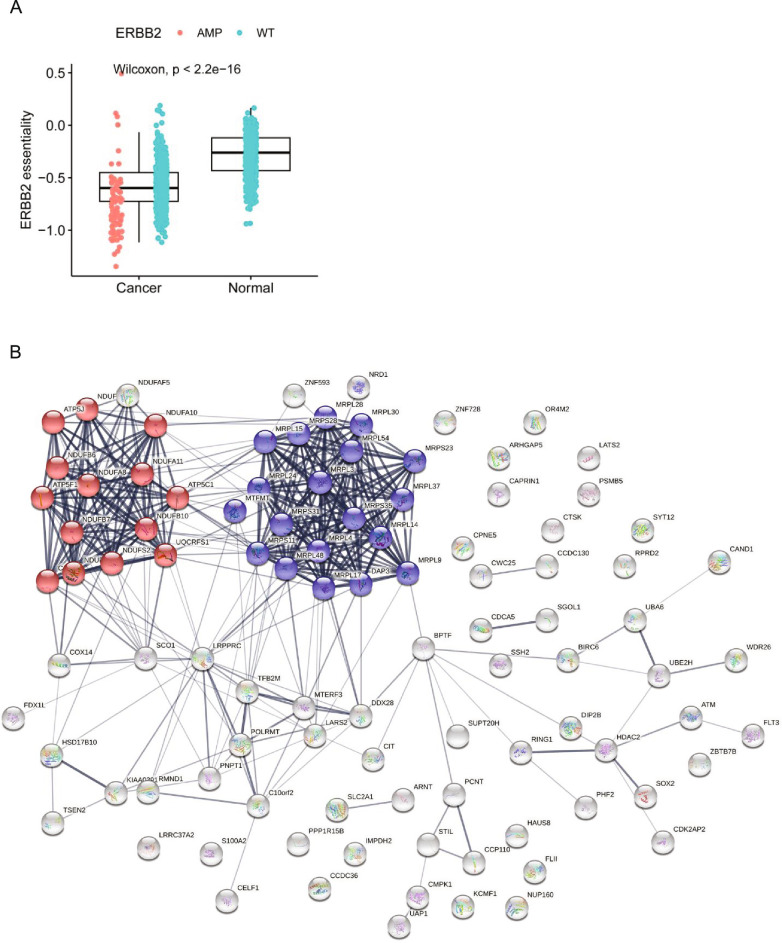
Cancer dependency maps have accelerated the discovery of tumor vulnerabilities that can be exploited as drug targets when translatable to patients. The Cancer Genome Atlas (TCGA) is a compendium of 'maps' detailing the genetic, epigenetic and molecular changes that occur during the pathogenesis of cancer, yet it lacks a dependency map to translate gene essentiality in patient tumors. Here, we used machine learning to build translational dependency maps for patient tumors, which identified tumor vulnerabilities that predict drug responses and disease outcomes. A similar approach was used to map gene tolerability in healthy tissues to prioritize tumor vulnerabilities with the best therapeutic windows. A subset of patient-translatable synthetic lethalities were experimentally tested, including PAPSS1/PAPSS12 and CNOT7/CNOT78, which were validated in vitro and in vivo. Notably, PAPSS1 synthetic lethality was driven by collateral deletion of PAPSS2 with PTEN and was correlated with patient survival. Finally, the translational dependency map is provided as a web-based application for exploring tumor vulnerabilities.
© 2024. The Author(s).
Conflict of interest statement
All authors except M.J.F. are employees of AbbVie. M.J.F. was an employee of AbbVie at the time of the study and is currently a full-time employee of Pfizer. The design, study conduct and financial support for this research were provided by AbbVie. AbbVie participated in the interpretation of data, review and approval of the publication.
Figures
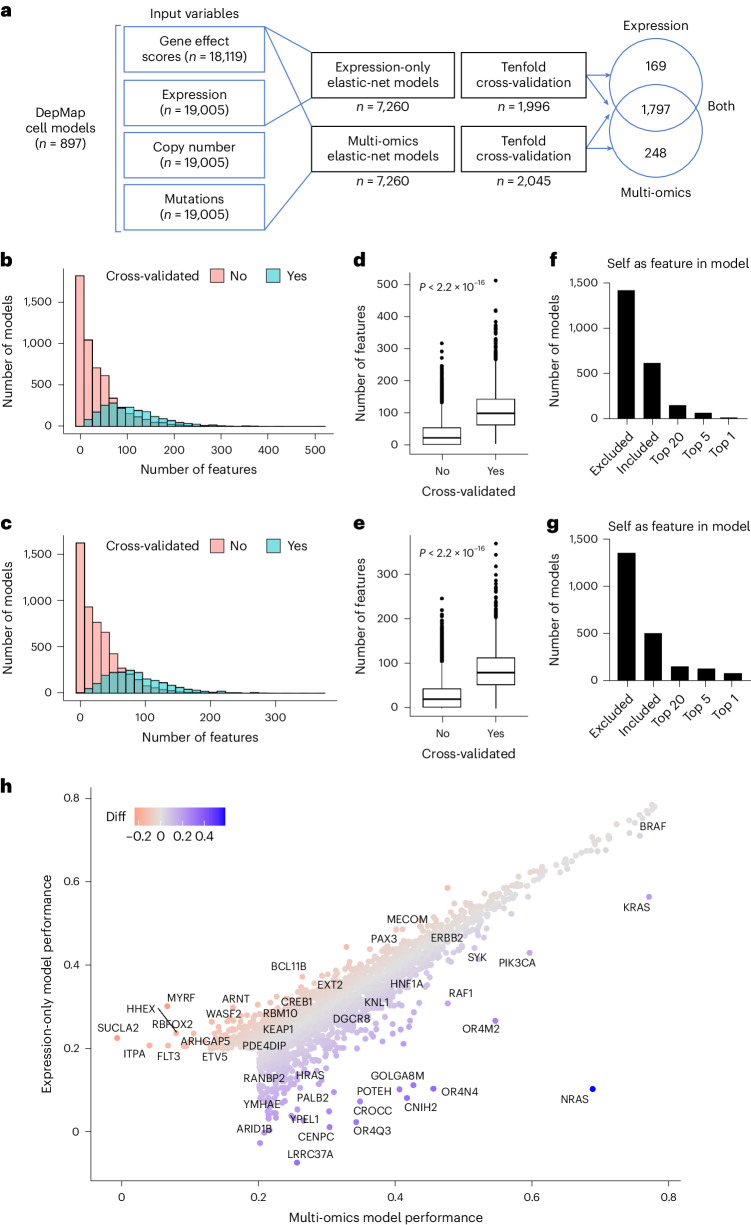
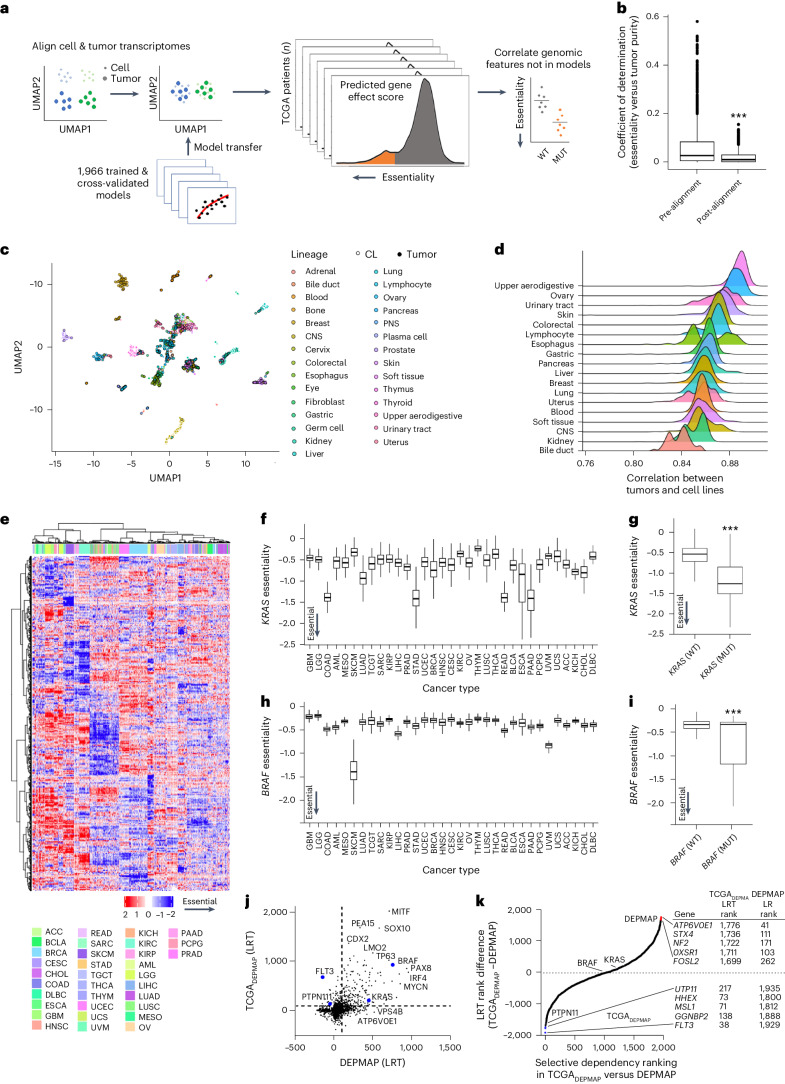
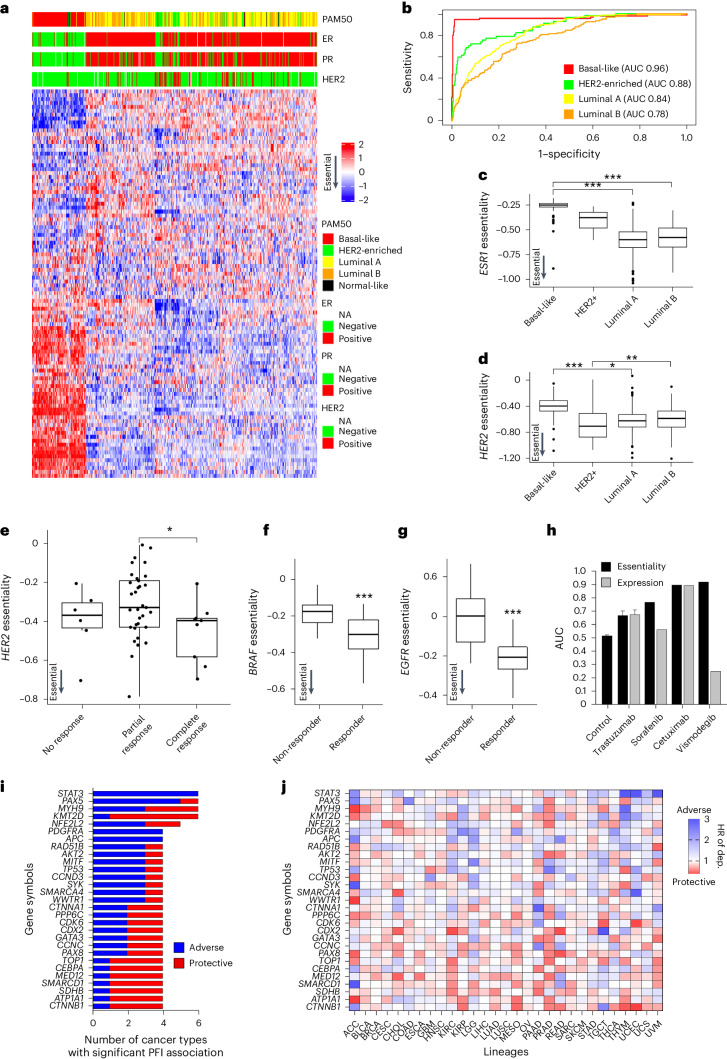
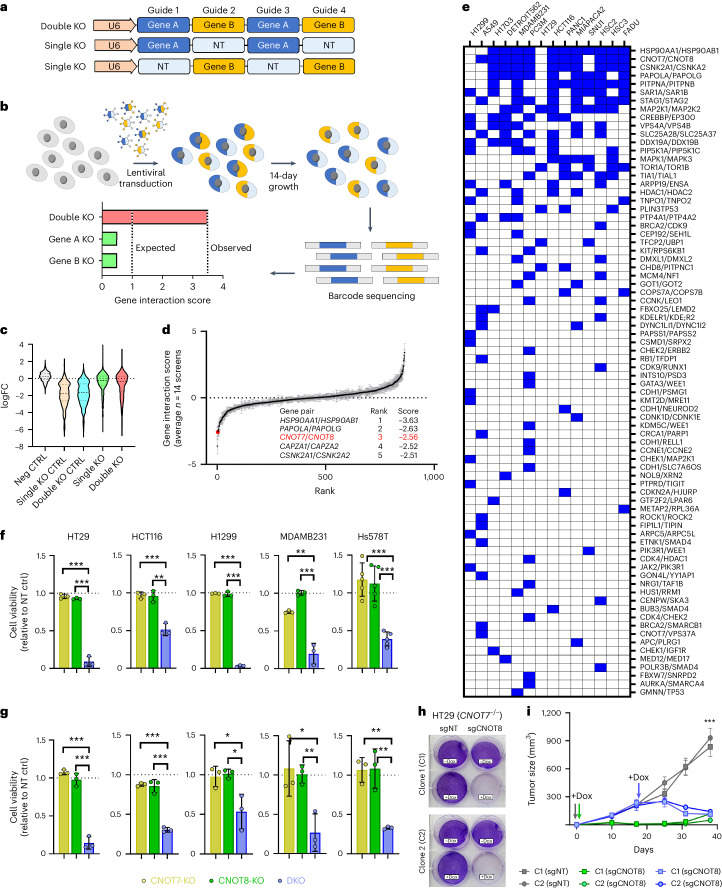
References
-
- Hutter, C. & Zenklusen, J. C. The Cancer Genome Atlas: creating lasting value beyond its data. Cell173, 283–285 (2018). - PubMed
-
- Srivastava, S., Ghosh, S., Kagan, J. & Mazurchuk, R. The PreCancer Atlas (PCA). Trends Cancer4, 513–514 (2018). - PubMed
-
- Sidaway, P. Tracing evolution reveals new biomarkers. Nat. Rev. Clin. Oncol.17, 5 (2020). - PubMed
-
- Haar, J. et al. Limited evolution of the actionable metastatic cancer genome under therapeutic pressure. Nat. Med.27, 1553–1563 (2021). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
