

Proteome-scale characterisation of motif-based interactome rewiring by disease mutations
- PMID: 39009827
- PMCID: PMC11369174
- DOI: 10.1038/s44320-024-00055-4
Proteome-scale characterisation of motif-based interactome rewiring by disease mutations
Abstract
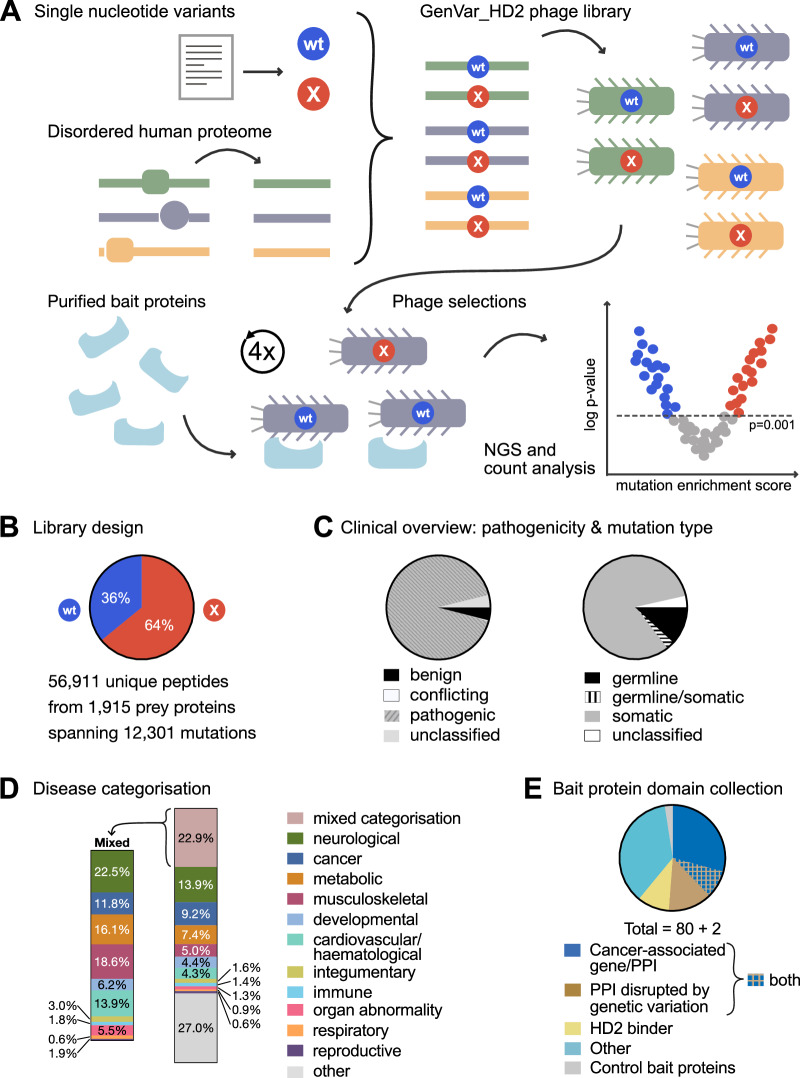
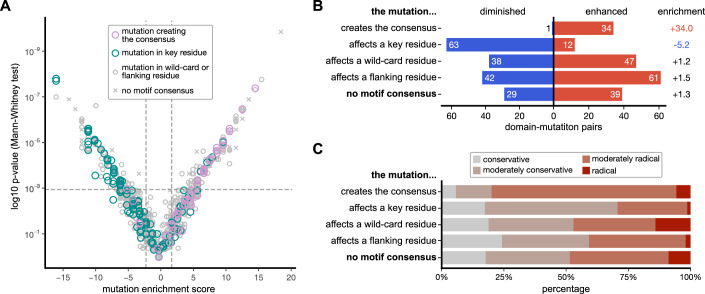
Whole genome and exome sequencing are reporting on hundreds of thousands of missense mutations. Taking a pan-disease approach, we explored how mutations in intrinsically disordered regions (IDRs) break or generate protein interactions mediated by short linear motifs. We created a peptide-phage display library tiling ~57,000 peptides from the IDRs of the human proteome overlapping 12,301 single nucleotide variants associated with diverse phenotypes including cancer, metabolic diseases and neurological diseases. By screening 80 human proteins, we identified 366 mutation-modulated interactions, with half of the mutations diminishing binding, and half enhancing binding or creating novel interaction interfaces. The effects of the mutations were confirmed by affinity measurements. In cellular assays, the effects of motif-disruptive mutations were validated, including loss of a nuclear localisation signal in the cell division control protein CDC45 by a mutation associated with Meier-Gorlin syndrome. The study provides insights into how disease-associated mutations may perturb and rewire the motif-based interactome.
Keywords: CDC45; Genetic Variation; Phage Display; Protein–Protein Interaction; Short Linear Motif.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
