

The use of constrained methods to analyze the molecular reactivity and to define a new type of pseudo atoms
- PMID: 39012379
- PMCID: PMC11252233
- DOI: 10.1007/s00894-024-06071-3
The use of constrained methods to analyze the molecular reactivity and to define a new type of pseudo atoms
Abstract
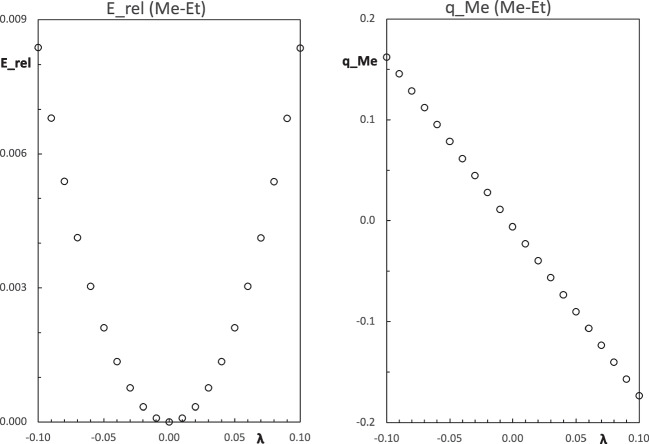
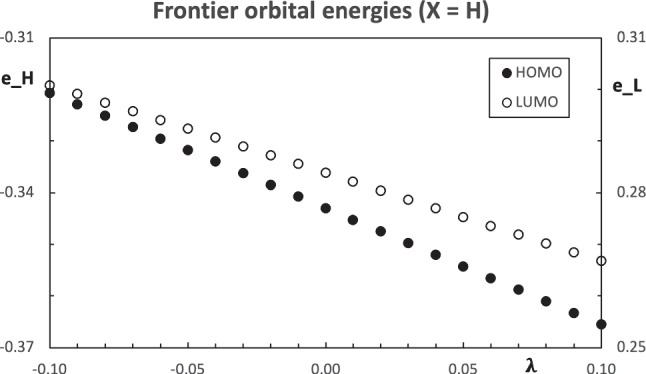
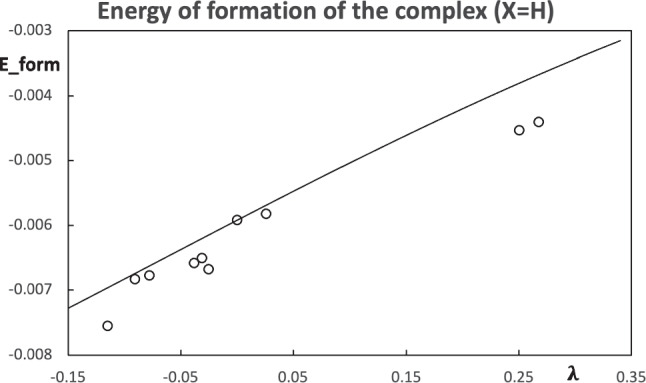
Context: Constrained methods in electronic structure methodologies add terms to the variational equations and generate solutions that represent distorted electronic distributions. In some cases, the new solutions can be used to study the chemical reactivity of parts of the molecule. Additionally, this contribution presents the use of population constraints to define pseudo atoms in a molecule. The effects of the pseudo atom on the molecular properties are analyzed. The pseudo atoms are used to simulate the inductive effect of the substituent in a group of carbonyl molecules and their effect on the stability of the complexes between these organic species and one molecule of water. A discussion on the assumptions involved in the present definition of pseudo atoms is also included.
Method: The constrained RHF computations are done in a modified Hartree-Fock code for Gaussian basis sets. The selected basis set is STO-6 G.
Keywords: Chemical reactivity; Constrained methods; Electronic structure; Hydrogen bonds; Pseudo atoms.
© 2024. The Author(s).
Figures








References
-
- Szabo A, Ostlund NS (1989) Modern quantum chemistry. McGraw-Hill, New York
-
- Parr R, Yang W (1989) Density functional theory of atoms and molecules. Oxford, Oxford
-
- Gross EKU, Dreizler R (1993) Density functional theory. Springer, Berlin
LinkOut - more resources
Full Text Sources
