

Investigation of CFTR Function in Human Nasal Epithelial Cells Informs Personalized Medicine
- PMID: 39012815
- PMCID: PMC11568479
- DOI: 10.1165/rcmb.2023-0398OC
Investigation of CFTR Function in Human Nasal Epithelial Cells Informs Personalized Medicine
Abstract
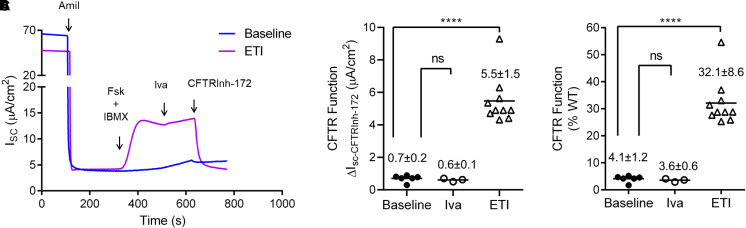
We broaden the clinical versatility of human nasal epithelial (HNE) cells. HNEs were isolated from 10 participants harboring cystic fibrosis transmembrane conductance regulator (CFTR) variants: 9 with rare variants (Q359R [n = 2], G480S, R334W [n = 5], and R560T) and 1 harboring R117H;7T;TG10/5T;TG12. Cultures were differentiated at the air-liquid interface. CFTR function was measured in Ussing chambers at three conditions: baseline, ivacaftor, and elexacaftor + tezacaftor + ivacaftor (ETI). Four participants initiated modulators. Q359R HNEs had 5.4% (% wild-type) baseline CFTR function and 25.5% with ivacaftor. With therapy, sweat [Cl-] decreased and symptoms resolved. G480S HNEs had 4.1% baseline and 32.1% CFTR function with ETI. Clinically, forced expiratory volume in 1 second increased and sweat [Cl-] decreased (119 to 46 mmol/L) with ETI. In vitro cultures derived from 5 participants harboring R334W showed a moderate increase in CFTR function with exposure to modulators. For one of these participants, ETI was begun in vivo; symptoms and forced expiratory volume in 1 second improved. The c.1679G>C (R560T) HNEs had less than 4% baseline CFTR function and no modulator response. RNA analysis confirmed that c.1679G>C completely missplices. A symptomatic patient harboring R117H;7T;TG10/5T;TG12 exhibited reduced CFTR function (17.5%) in HNEs, facilitating a diagnosis of mild CF. HNEs responded to modulators (ivacaftor: 32.8%, ETI: 55.5%), and, since beginning therapy, lung function improved. We reaffirm HNE use for guiding therapeutic approaches, inform predictions on modulator response (e.g., R334W), and closely assess variants that affect splicing (e.g., c.1679G>C). Notably, functional studies in HNEs harboring R117H;7T;TG10/5T;TG12 facilitated a diagnosis of mild CF, suggesting the use for HNE functional studies as a clinical diagnostic test.
Keywords: 5T; air–liquid interface; cystic fibrosis; modulators; nasal epithelial.
Figures





References
-
- Noel S, Servel N, Hatton A, Golec A, Rodrat M, Ng DRS, et al. Correlating genotype with phenotype using CFTR-mediated whole-cell Cl(−) currents in human nasal epithelial cells. J Physiol . 2022;600:1515–1531. - PubMed
-
- Amaral MD, de Boeck K, CF ESPTFoSuatndf Theranostics by testing CFTR modulators in patient-derived materials: the current status and a proposal for subjects with rare CFTR mutations. J Cyst Fibros . 2019;18:685–692. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
