

Comprehensive EHMT1 variants analysis broadens genotype-phenotype associations and molecular mechanisms in Kleefstra syndrome
- PMID: 39013458
- PMCID: PMC11339614
- DOI: 10.1016/j.ajhg.2024.06.008
Comprehensive EHMT1 variants analysis broadens genotype-phenotype associations and molecular mechanisms in Kleefstra syndrome
Abstract
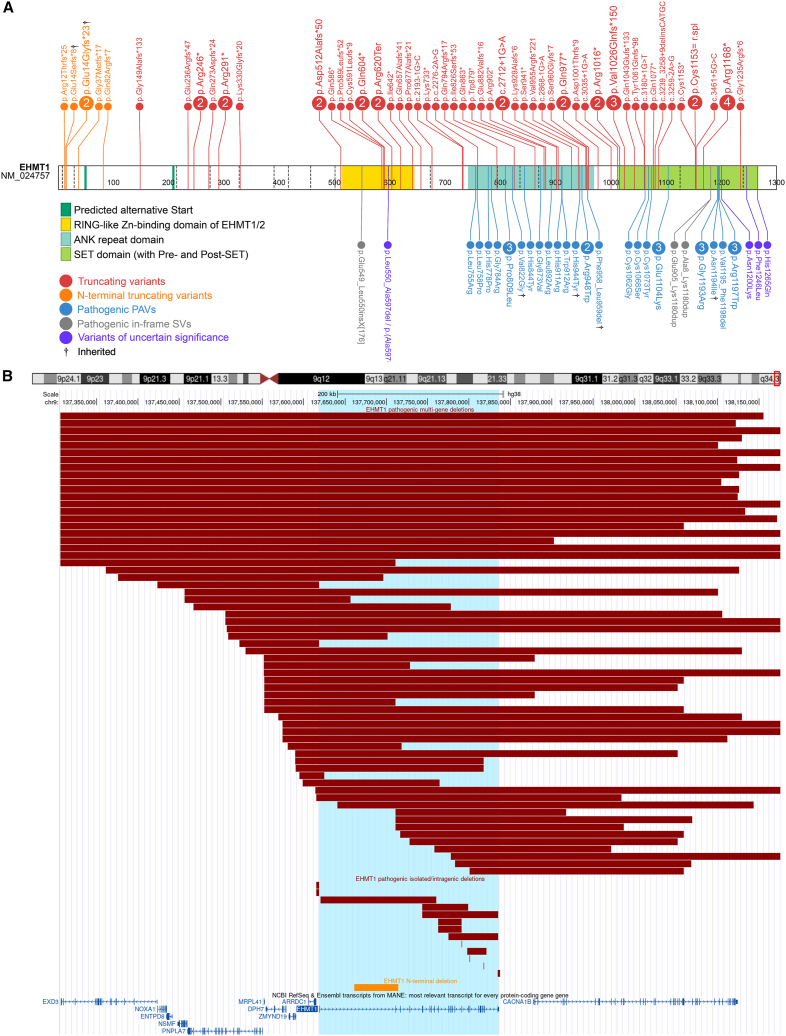
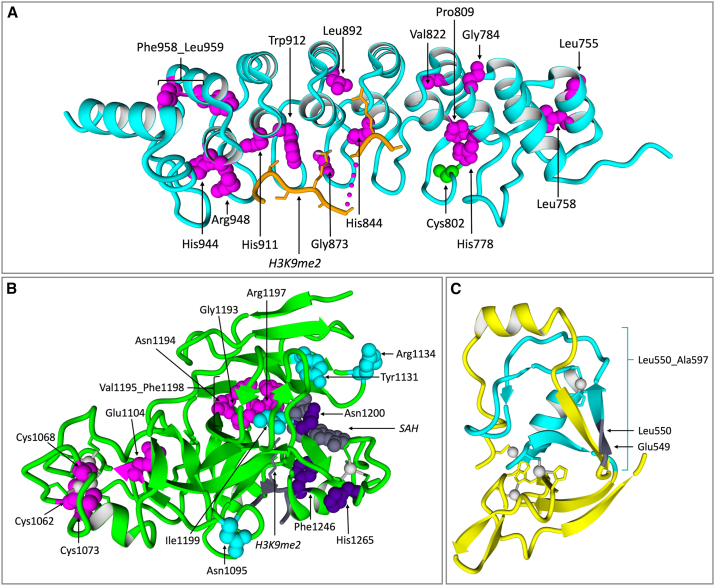
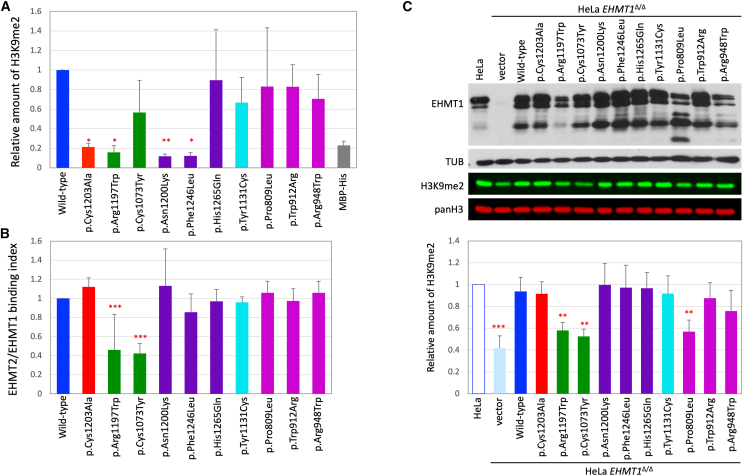
The shift to a genotype-first approach in genetic diagnostics has revolutionized our understanding of neurodevelopmental disorders, expanding both their molecular and phenotypic spectra. Kleefstra syndrome (KLEFS1) is caused by EHMT1 haploinsufficiency and exhibits broad clinical manifestations. EHMT1 encodes euchromatic histone methyltransferase-1-a pivotal component of the epigenetic machinery. We have recruited 209 individuals with a rare EHMT1 variant and performed comprehensive molecular in silico and in vitro testing alongside DNA methylation (DNAm) signature analysis for the identified variants. We (re)classified the variants as likely pathogenic/pathogenic (molecularly confirming Kleefstra syndrome) in 191 individuals. We provide an updated and broader clinical and molecular spectrum of Kleefstra syndrome, including individuals with normal intelligence and familial occurrence. Analysis of the EHMT1 variants reveals a broad range of molecular effects and their associated phenotypes, including distinct genotype-phenotype associations. Notably, we showed that disruption of the "reader" function of the ankyrin repeat domain by a protein altering variant (PAV) results in a KLEFS1-specific DNAm signature and milder phenotype, while disruption of only "writer" methyltransferase activity of the SET domain does not result in KLEFS1 DNAm signature or typical KLEFS1 phenotype. Similarly, N-terminal truncating variants result in a mild phenotype without the DNAm signature. We demonstrate how comprehensive variant analysis can provide insights into pathogenesis of the disorder and DNAm signature. In summary, this study presents a comprehensive overview of KLEFS1 and EHMT1, revealing its broader spectrum and deepening our understanding of its molecular mechanisms, thereby informing accurate variant interpretation, counseling, and clinical management.
Keywords: DNA methylation; EHMT1; H3K9; Kleefstra syndrome; NDD; neurodevelopmental disorders.
Copyright © 2024 American Society of Human Genetics. All rights reserved.
Conflict of interest statement
Declaration of interests A.M.M. is an employee of GeneDx, LLC. B.S. is a shareholder in EpiSign Inc., a biotechnology company involved in commercialization of EpiSign technology.
Figures

References
-
- Wilczewski C.M., Obasohan J., Paschall J.E., Zhang S., Singh S., Maxwell G.L., Similuk M., Wolfsberg T.G., Turner C., Biesecker L.G., Katz A.E. Genotype first: Clinical genomics research through a reverse phenotyping approach. Am. J. Hum. Genet. 2023;110:3–12. doi: 10.1016/j.ajhg.2022.12.004. - DOI - PMC - PubMed
-
- Rots D., Chater-Diehl E., Dingemans A.J.M., Goodman S.J., Siu M.T., Cytrynbaum C., Choufani S., Hoang N., Walker S., Awamleh Z., et al. Truncating SRCAP variants outside the Floating-Harbor syndrome locus cause a distinct neurodevelopmental disorder with a specific DNA methylation signature. Am. J. Hum. Genet. 2021;108:1053–1068. doi: 10.1016/j.ajhg.2021.04.008. - DOI - PMC - PubMed
-
- Cuvertino S., Hartill V., Colyer A., Garner T., Nair N., Al-Gazali L., Canham N., Faundes V., Flinter F., Hertecant J., et al. A restricted spectrum of missense KMT2D variants cause a multiple malformations disorder distinct from Kabuki syndrome. Genet. Med. 2020;22:867–877. doi: 10.1038/s41436-019-0743-3. - DOI - PMC - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
