

Linking LRP12 CGG repeat expansion to inherited peripheral neuropathy
- PMID: 39013564
- PMCID: PMC11877035
- DOI: 10.1136/jnnp-2024-333403
Linking LRP12 CGG repeat expansion to inherited peripheral neuropathy
Abstract
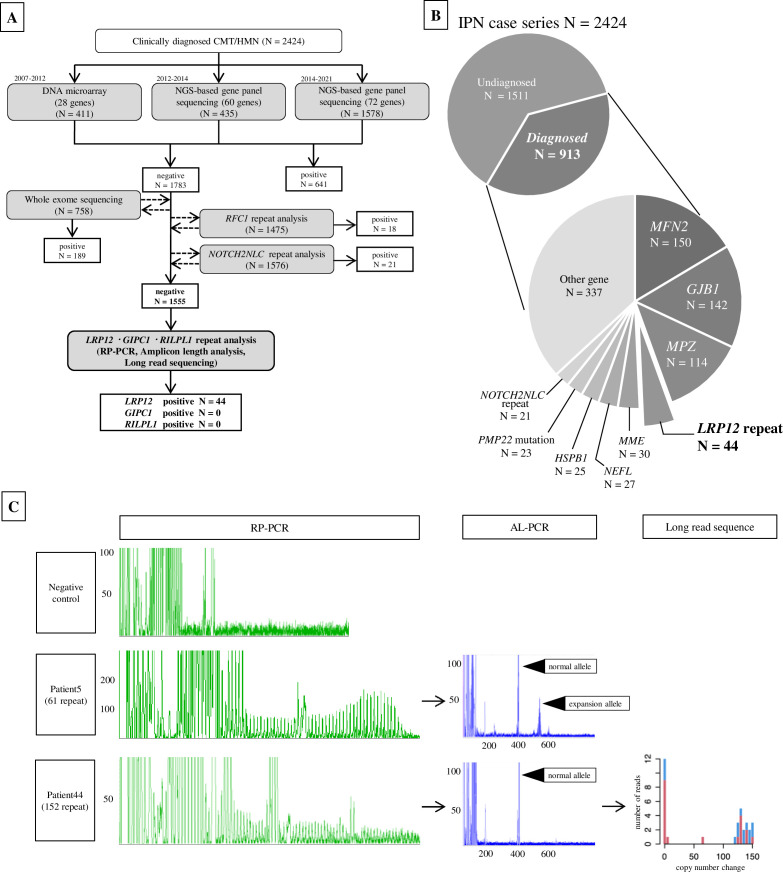
Background: The causative genes for over 60% of inherited peripheral neuropathy (IPN) remain unidentified. This study endeavours to enhance the genetic diagnostic rate in IPN cases by conducting screenings focused on non-coding repeat expansions.
Methods: We gathered data from 2424 unrelated Japanese patients diagnosed with IPN, among whom 1555 cases with unidentified genetic causes, as determined through comprehensive prescreening analyses, were selected for the study. Screening for CGG non-coding repeat expansions in LRP12, GIPC1 and RILPL1 genes was conducted using PCR and long-read sequencing technologies.
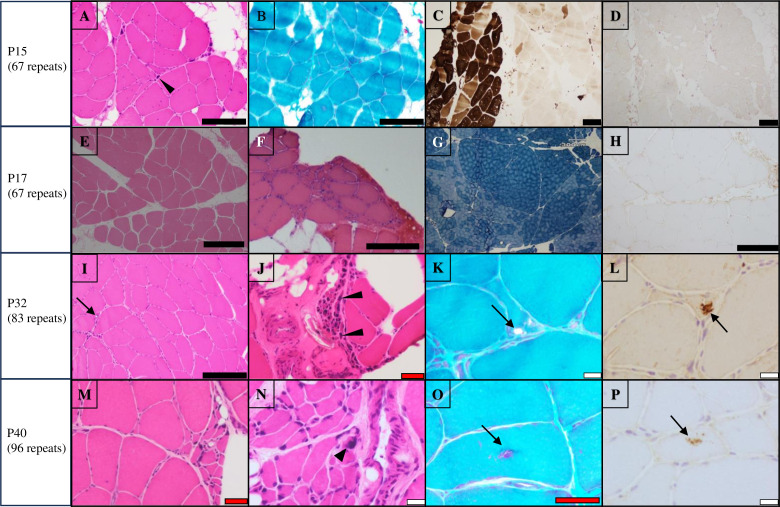
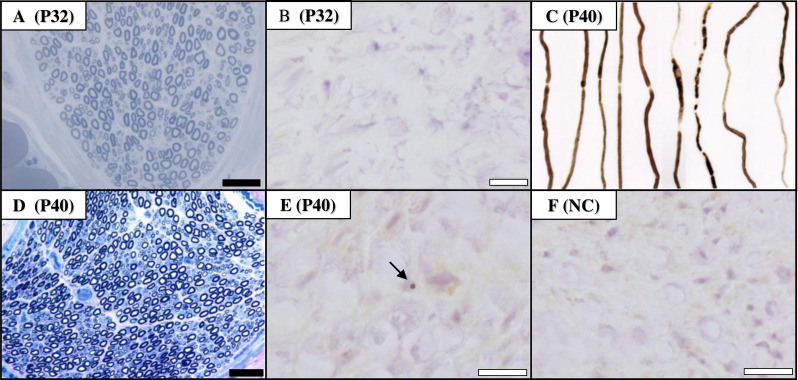
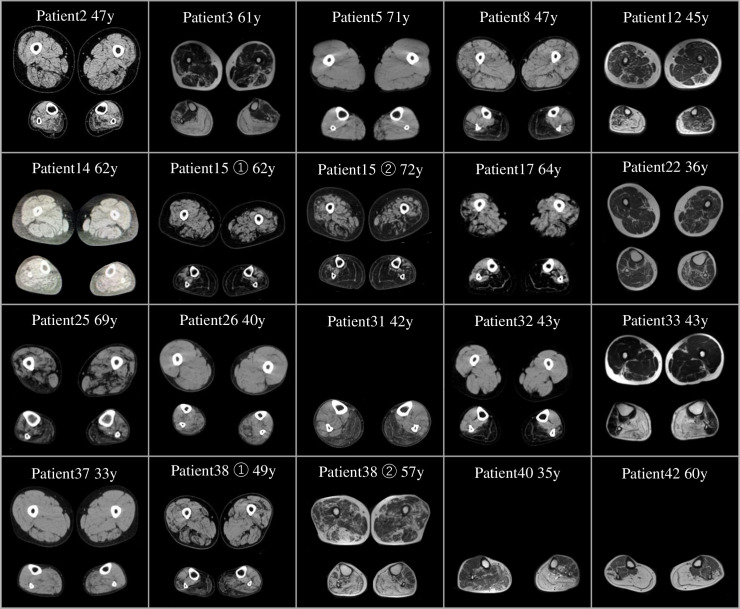
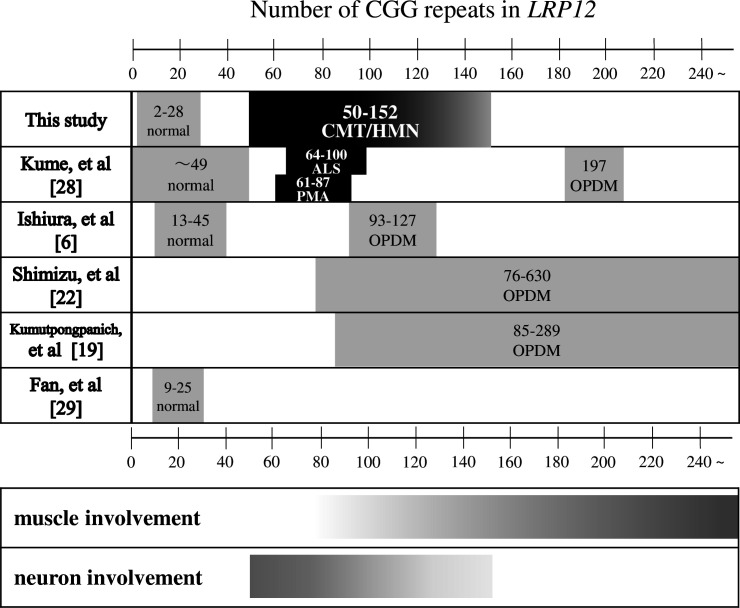
Results: We identified CGG repeat expansions in LRP12 from 44 cases, establishing it as the fourth most common aetiology in Japanese IPN. Most cases (29/37) exhibited distal limb weakness, without ptosis, ophthalmoplegia, facial muscle weakness or bulbar palsy. Neurogenic changes were frequently observed in both needle electromyography (97%) and skeletal muscle tissue (100%). In nerve conduction studies, 28 cases primarily showed impairment in motor nerves without concurrent involvement of sensory nerves, consistent with the phenotype of hereditary motor neuropathy. In seven cases, both motor and sensory nerves were affected, resembling the Charcot-Marie-Tooth (CMT) phenotype. Importantly, the mean CGG repeat number detected in the present patients was significantly shorter than that of patients with LRP12-oculopharyngodistal myopathy (p<0.0001). Additionally, GIPC1 and RILPL1 repeat expansions were absent in our IPN cases.
Conclusion: We initially elucidate LRP12 repeat expansions as a prevalent cause of CMT, highlighting the necessity for an adapted screening strategy in clinical practice, particularly when addressing patients with IPN.
Keywords: HMSN (CHARCOT-MARIE-TOOTH); NEUROGENETICS; NEUROMUSCULAR; NEUROPATHY.
© Author(s) (or their employer(s)) 2025. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ Group.
Conflict of interest statement
Competing interests: None declared.
Figures
References
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical