

Haplotype-aware sequence alignment to pangenome graphs
- PMID: 39013594
- PMCID: PMC11529843
- DOI: 10.1101/gr.279143.124
Haplotype-aware sequence alignment to pangenome graphs
Abstract
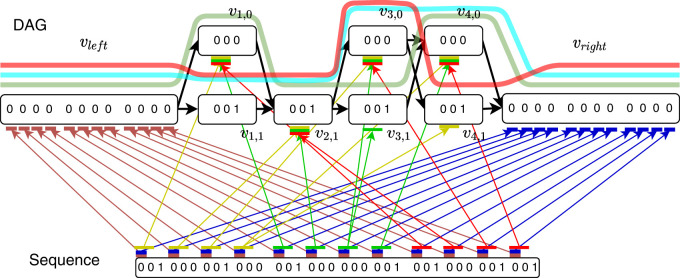
Modern pangenome graphs are built using haplotype-resolved genome assemblies. When mapping reads to a pangenome graph, prioritizing alignments that are consistent with the known haplotypes improves genotyping accuracy. However, the existing rigorous formulations for colinear chaining and alignment problems do not consider the haplotype paths in a pangenome graph. This often leads to spurious read alignments to those paths that are unlikely recombinations of the known haplotypes. In this paper, we develop novel formulations and algorithms for sequence-to-graph alignment and chaining problems. Inspired by the genotype imputation models, we assume that a query sequence is an imperfect mosaic of reference haplotypes. Accordingly, we introduce a recombination penalty in the scoring functions for each haplotype switch. First, we solve haplotype-aware sequence-to-graph alignment in [Formula: see text] time, where Q is the query sequence, E is the set of edges, and H is the set of haplotypes represented in the graph. To complement our solution, we prove that an algorithm significantly faster than [Formula: see text] is impossible under the strong exponential time hypothesis (SETH). Second, we propose a haplotype-aware chaining algorithm that runs in [Formula: see text] time after graph preprocessing, where N is the count of input anchors. We then establish that a chaining algorithm significantly faster than [Formula: see text] is impossible under SETH. As a proof-of-concept, we implemented our chaining algorithm in the Minichain aligner. By aligning sequences sampled from the human major histocompatibility complex (MHC) to a pangenome graph of 60 MHC haplotypes, we demonstrate that our algorithm achieves better consistency with ground-truth recombinations compared with a haplotype-agnostic algorithm.
© 2024 Chandra et al.; Published by Cold Spring Harbor Laboratory Press.
Figures






References
-
- Abouelhoda M, Ohlebusch E. 2005. Chaining algorithms for multiple genome comparison. J Discrete Algorithms 3: 321–341. 10.1016/j.jda.2004.08.011 - DOI
-
- Amir A, Lewenstein M, Lewenstein N. 2000. Pattern matching in hypertext. J Algorithms 35: 82–99. 10.1006/jagm.1999.1063 - DOI
-
- Backurs A, Indyk P. 2015. Edit distance cannot be computed in strongly subquadratic time (unless SETH is false). In Proceedings of the Forty-Seventh Annual ACM Symposium on Theory of Computing, Portland, OR, pp. 51–58. Association for Computing Machinery.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials