

GRAIL1 Stabilizes Misfolded Mutant p53 through a Ubiquitin Ligase-Independent, Chaperone Regulatory Function
- PMID: 39018356
- PMCID: PMC11530312
- DOI: 10.1158/1541-7786.MCR-24-0361
GRAIL1 Stabilizes Misfolded Mutant p53 through a Ubiquitin Ligase-Independent, Chaperone Regulatory Function
Abstract
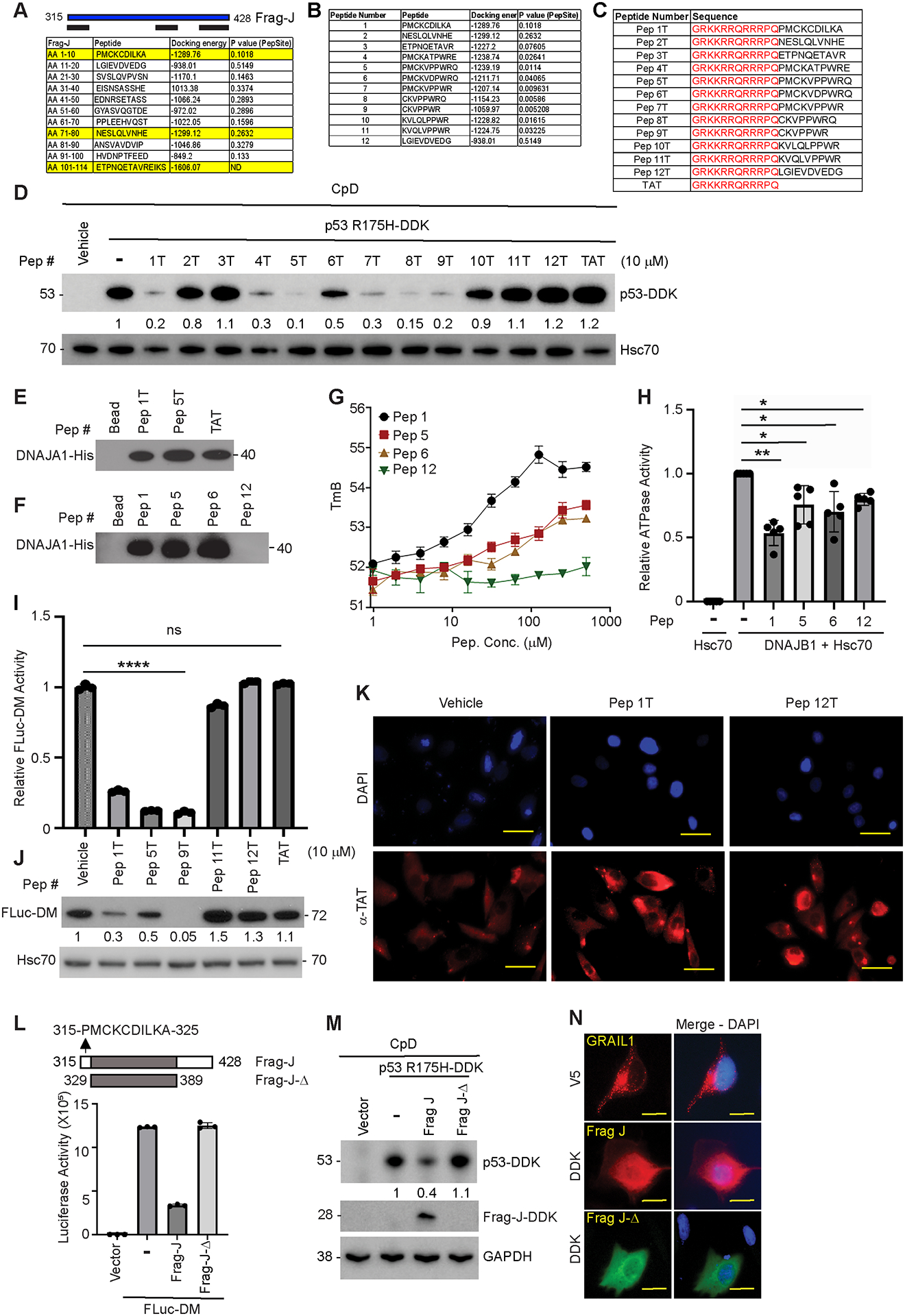
Frequent (>70%) TP53 mutations often promote its protein stabilization, driving esophageal adenocarcinoma (EAC) development linked to poor survival and therapy resistance. We previously reported that during Barrett's esophagus progression to EAC, an isoform switch occurs in the E3 ubiquitin ligase RNF128 (aka GRAIL-gene related to anergy in lymphocytes), enriching isoform 1 (hereby GRAIL1) and stabilizing the mutant p53 protein. Consequently, GRAIL1 knockdown degrades mutant p53. But, how GRAIL1 stabilizes the mutant p53 protein remains unclear. In search for a mechanism, here, we performed biochemical and cell biology studies to identify that GRAIL has a binding domain (315-PMCKCDILKA-325) for heat shock protein 40/DNAJ. This interaction can influence DNAJ chaperone activity to modulate misfolded mutant p53 stability. As predicted, either the overexpression of a GRAIL fragment (Frag-J) encompassing the DNAJ binding domain or a cell-permeable peptide (Pep-J) encoding the above 10 amino acids can bind and inhibit DNAJ-Hsp70 co-chaperone activity, thus degrading misfolded mutant p53. Consequently, either Frag-J or Pep-J can reduce the survival of mutant p53 containing dysplastic Barrett's esophagus and EAC cells and inhibit the growth of patient-derived organoids of dysplastic Barrett's esophagus in 3D cultures. The misfolded mutant p53 targeting and growth inhibitory effects of Pep-J are comparable with simvastatin, a cholesterol-lowering drug that can degrade misfolded mutant p53 also via inhibiting DNAJA1, although by a distinct mechanism. Implications: We identified a novel ubiquitin ligase-independent, chaperone-regulating domain in GRAIL and further synthesized a first-in-class novel misfolded mutant p53 degrading peptide having future translational potential.
©2024 American Association for Cancer Research.
Conflict of interest statement
Figures






References
-
- Ray P, Nancarrow DJ, Ferrer-Torres D, Wang Z, San Martinho M, Hinton T, et al. UBCH5 Family Members Differentially Impact Stabilization of Mutant p53 via RNF128 Iso1 During Barrett’s Progression to Esophageal Adenocarcinoma. Cellular and molecular gastroenterology and hepatology 2021. doi 10.1016/j.jcmgh.2021.08.003. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
