

Rare variation in non-coding regions with evolutionary signatures contributes to autism spectrum disorder risk
- PMID: 39019033
- PMCID: PMC11406188
- DOI: 10.1016/j.xgen.2024.100609
Rare variation in non-coding regions with evolutionary signatures contributes to autism spectrum disorder risk
Abstract
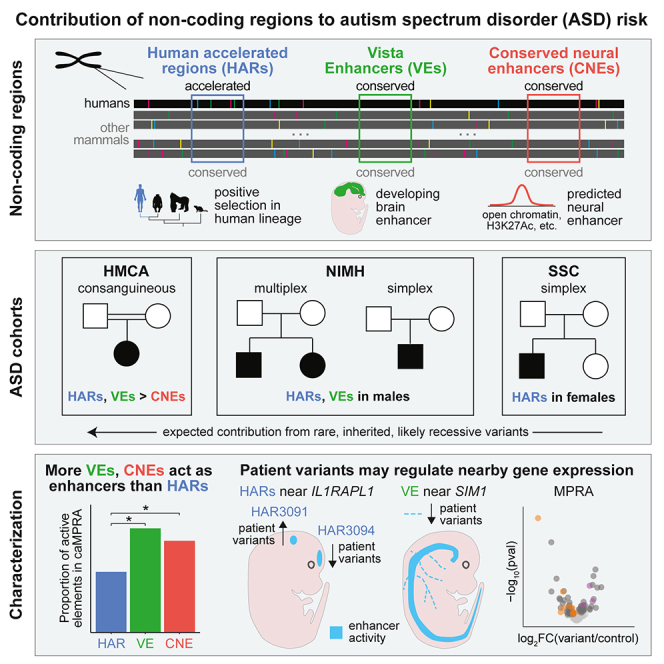
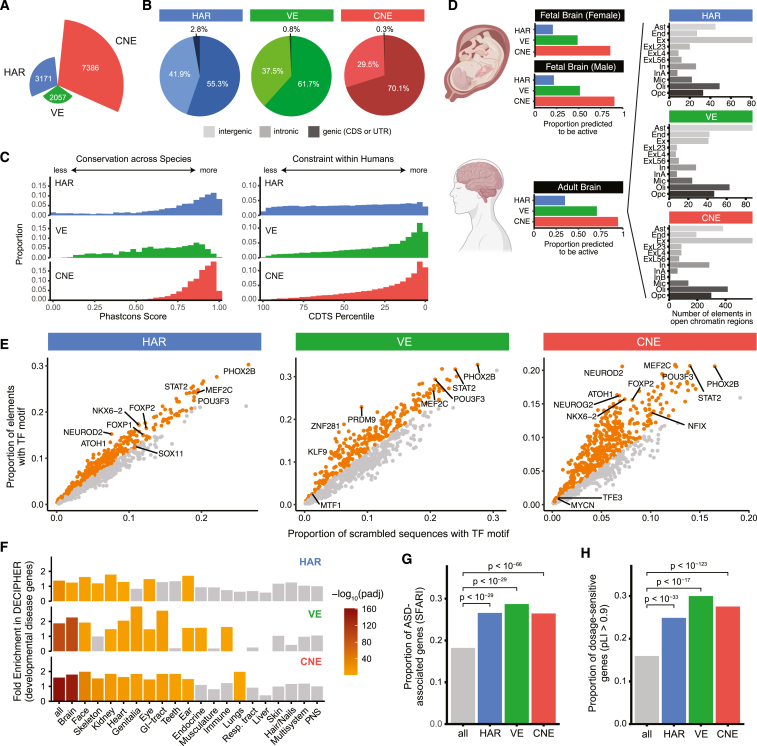
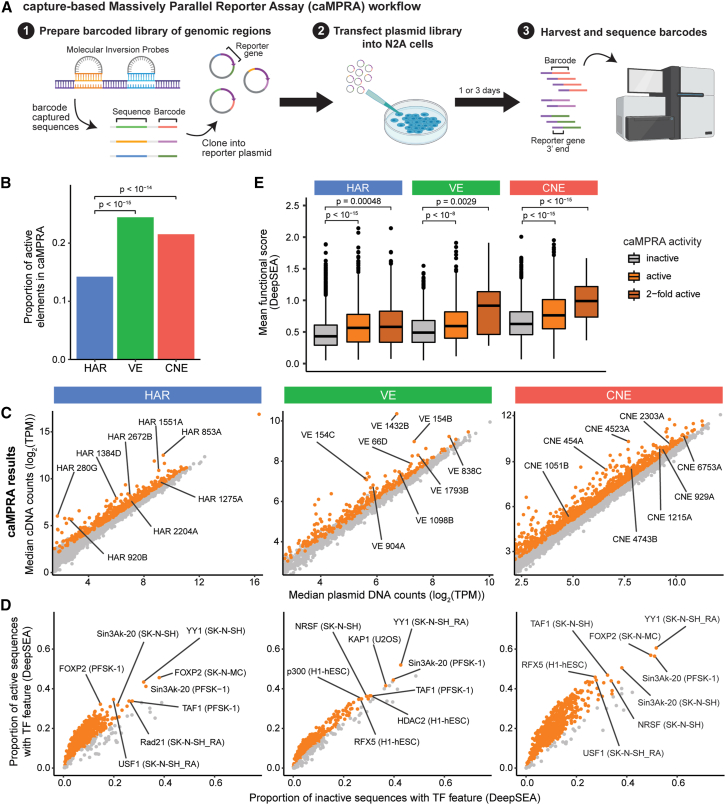
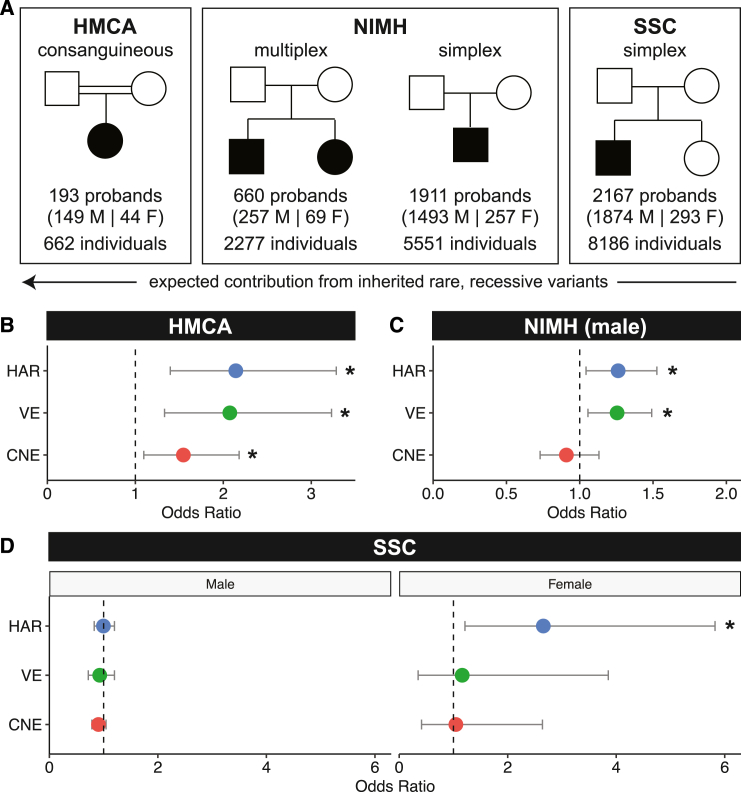
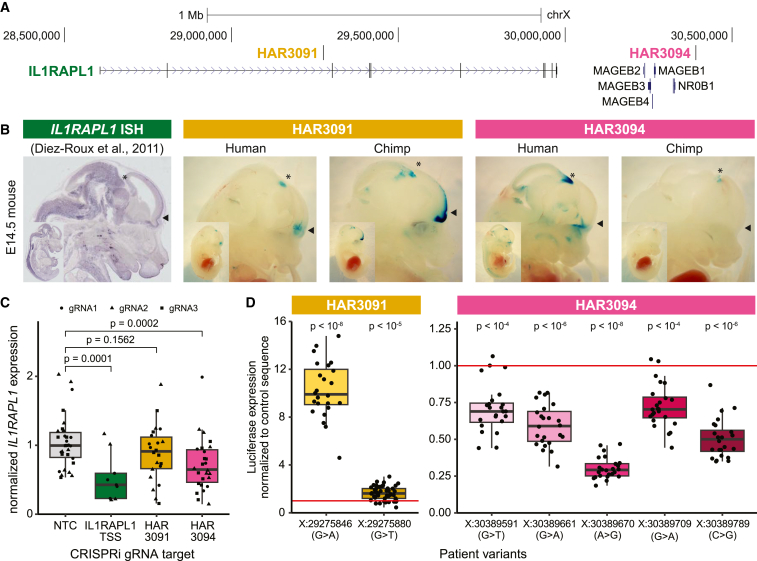
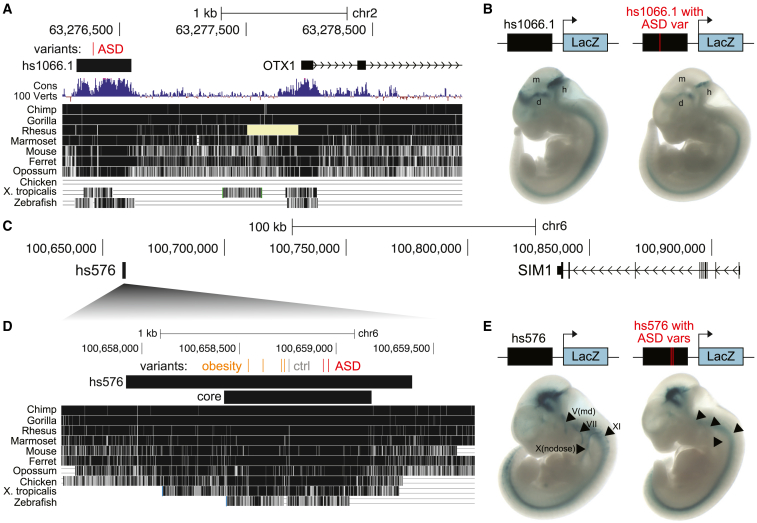
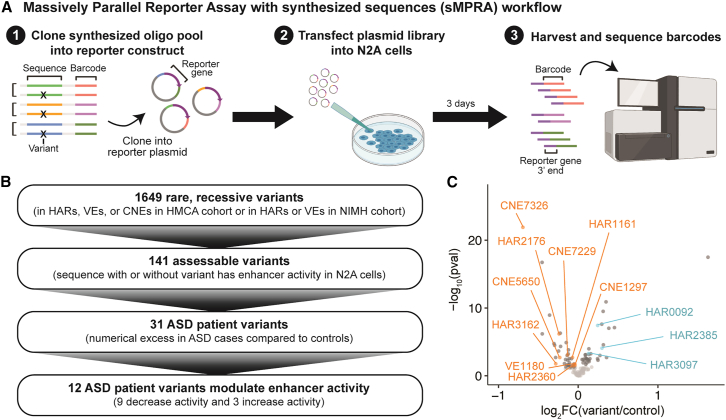
Little is known about the role of non-coding regions in the etiology of autism spectrum disorder (ASD). We examined three classes of non-coding regions: human accelerated regions (HARs), which show signatures of positive selection in humans; experimentally validated neural VISTA enhancers (VEs); and conserved regions predicted to act as neural enhancers (CNEs). Targeted and whole-genome analysis of >16,600 samples and >4,900 ASD probands revealed that likely recessive, rare, inherited variants in HARs, VEs, and CNEs substantially contribute to ASD risk in probands whose parents share ancestry, which enriches for recessive contributions, but modestly contribute, if at all, in simplex family structures. We identified multiple patient variants in HARs near IL1RAPL1 and in VEs near OTX1 and SIM1 and showed that they change enhancer activity. Our results implicate both human-evolved and evolutionarily conserved non-coding regions in ASD risk and suggest potential mechanisms of how regulatory changes can modulate social behavior.
Keywords: IL1RAPL1; OTX1; SIM1; VISTA enhancers; autism spectrum disorder; caMPRA; consanguineous families; conserved neural enhancers; human accelerated regions; noncoding regions.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
Update of
-
Rare variation in noncoding regions with evolutionary signatures contributes to autism spectrum disorder risk.medRxiv [Preprint]. 2023 Sep 22:2023.09.19.23295780. doi: 10.1101/2023.09.19.23295780. medRxiv. 2023. Update in: Cell Genom. 2024 Aug 14;4(8):100609. doi: 10.1016/j.xgen.2024.100609. PMID: 37790480 Free PMC article. Updated. Preprint.
References
-
- Maenner M.J., Warren Z., Williams A.R., Amoakohene E., Bakian A.V., Bilder D.A., Durkin M.S., Fitzgerald R.T., Furnier S.M., Hughes M.M., et al. Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2020. MMWR. Surveill. Summ. 2023;72:1–14. - PMC - PubMed
-
- Hyman S.L., Levy S.E., Myers S.M., COUNCIL ON CHILDREN WITH DISABILITIES, SECTION ON DEVELOPMENTAL AND BEHAVIORAL PEDIATRICS Identification, Evaluation, and Management of Children With Autism Spectrum Disorder. Pediatrics. 2020;145 - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
