

In vivo single-cell CRISPR uncovers distinct TNF programmes in tumour evolution
- PMID: 39020166
- PMCID: PMC11306103
- DOI: 10.1038/s41586-024-07663-y
In vivo single-cell CRISPR uncovers distinct TNF programmes in tumour evolution
Abstract
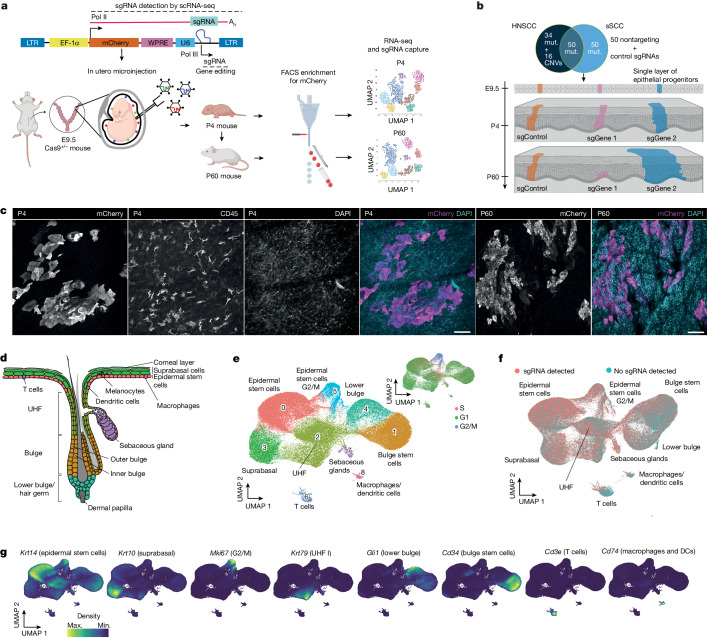
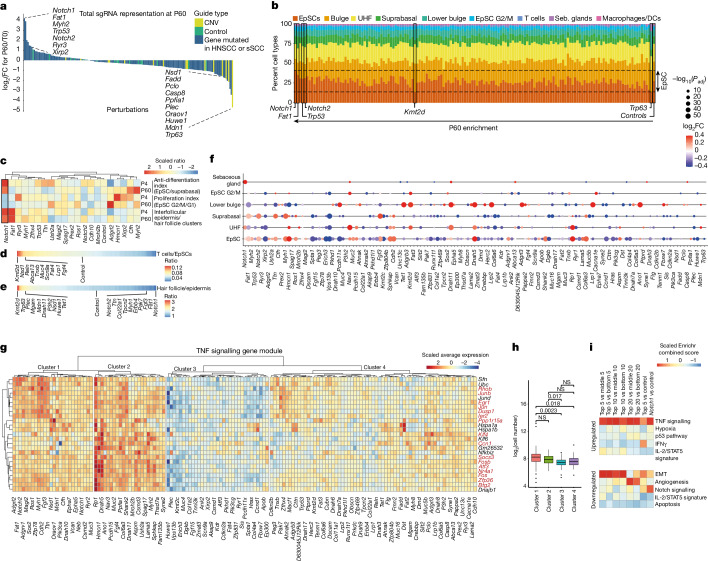
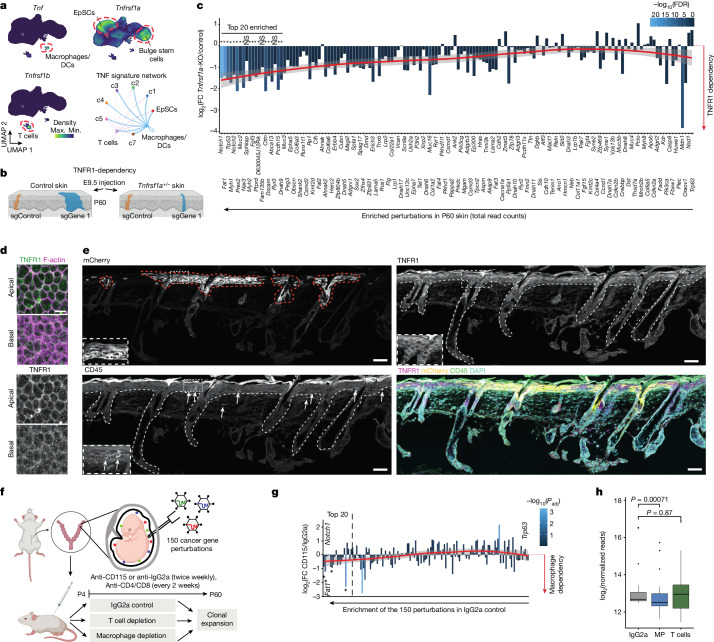
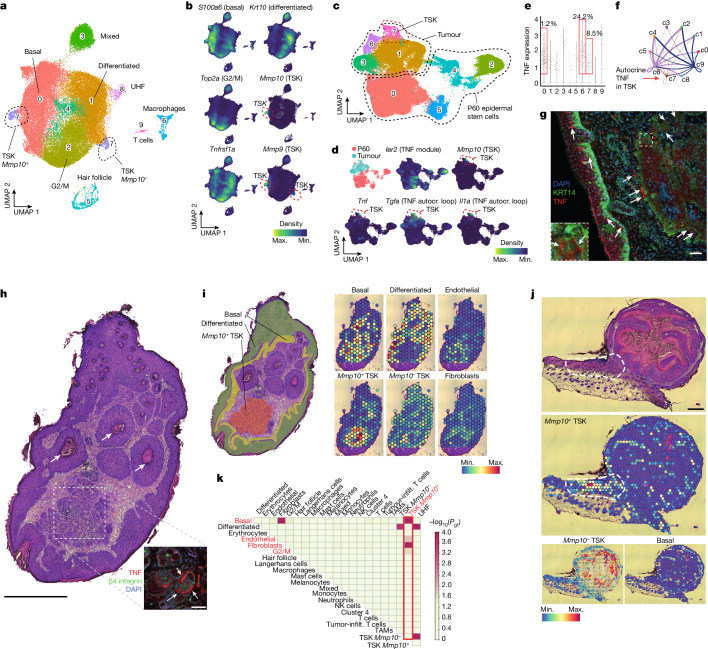
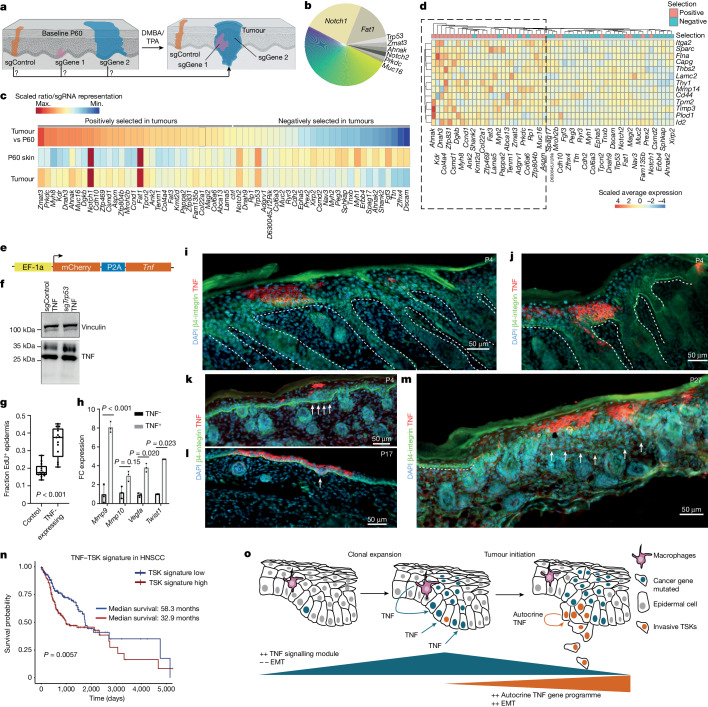
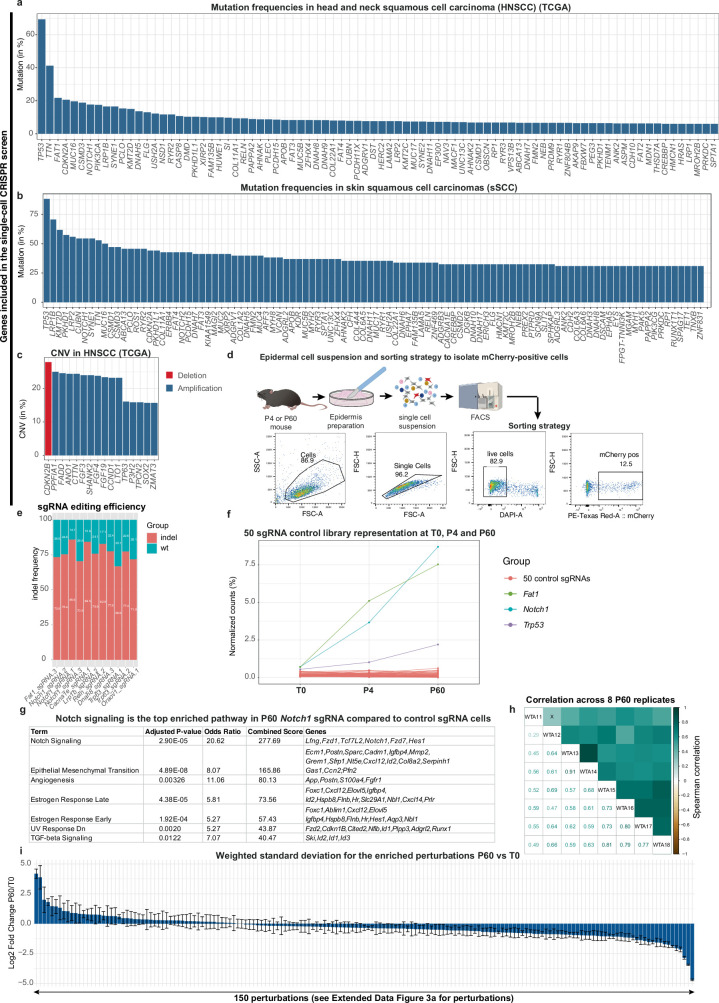
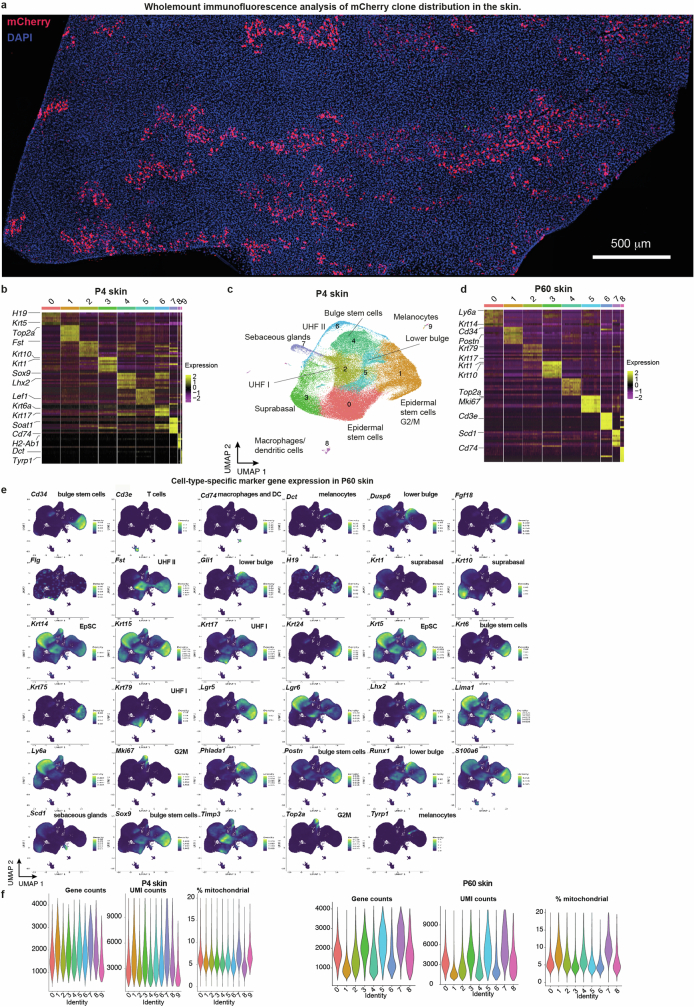
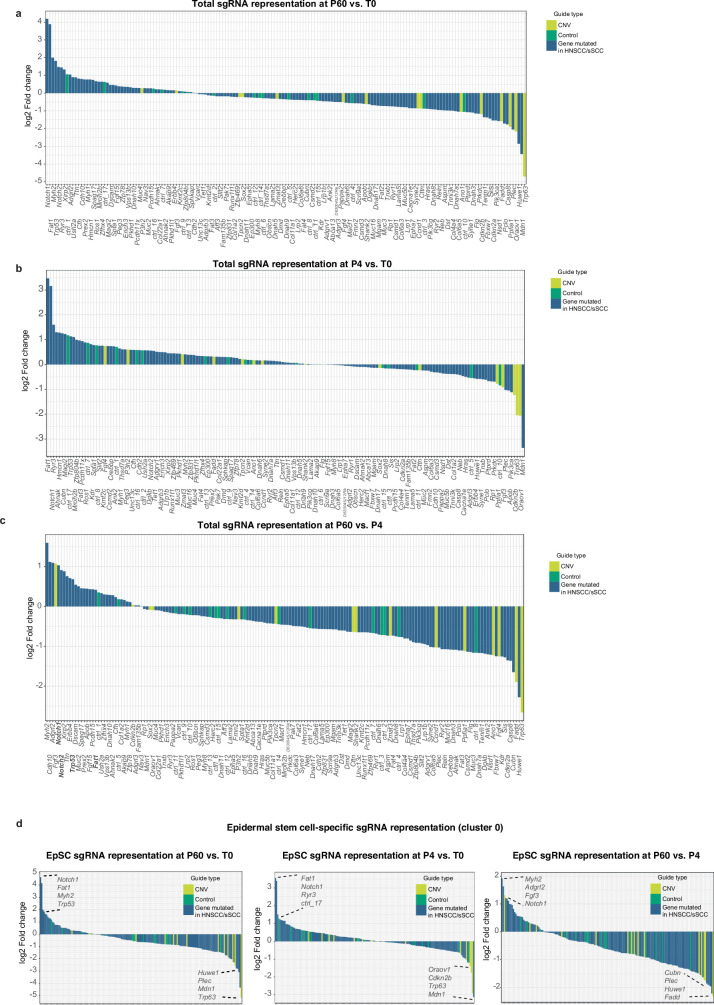
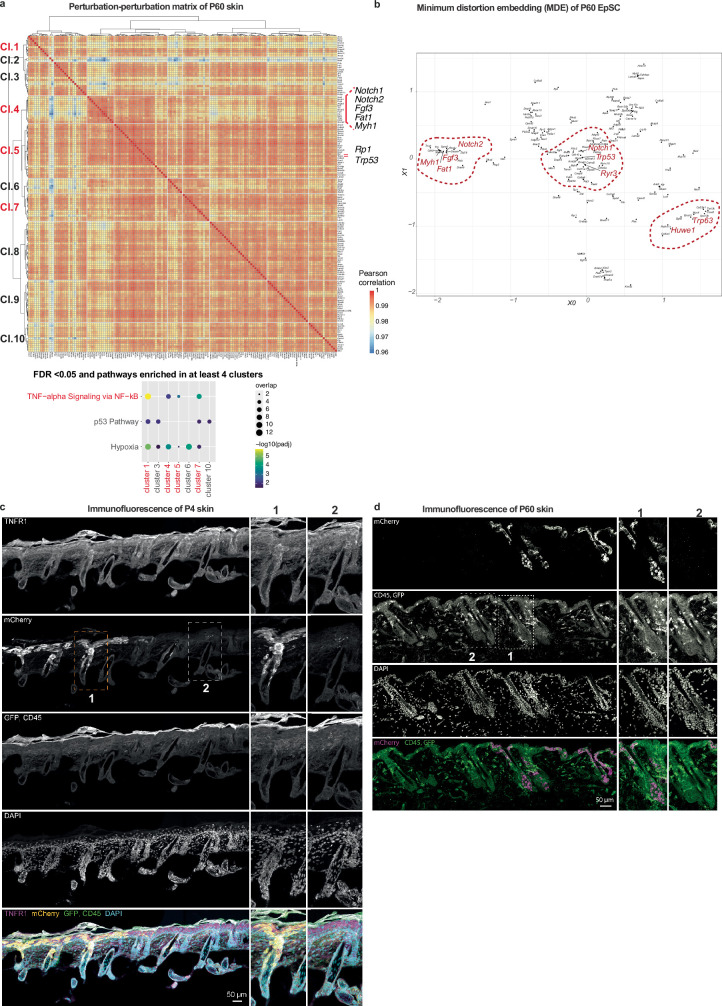
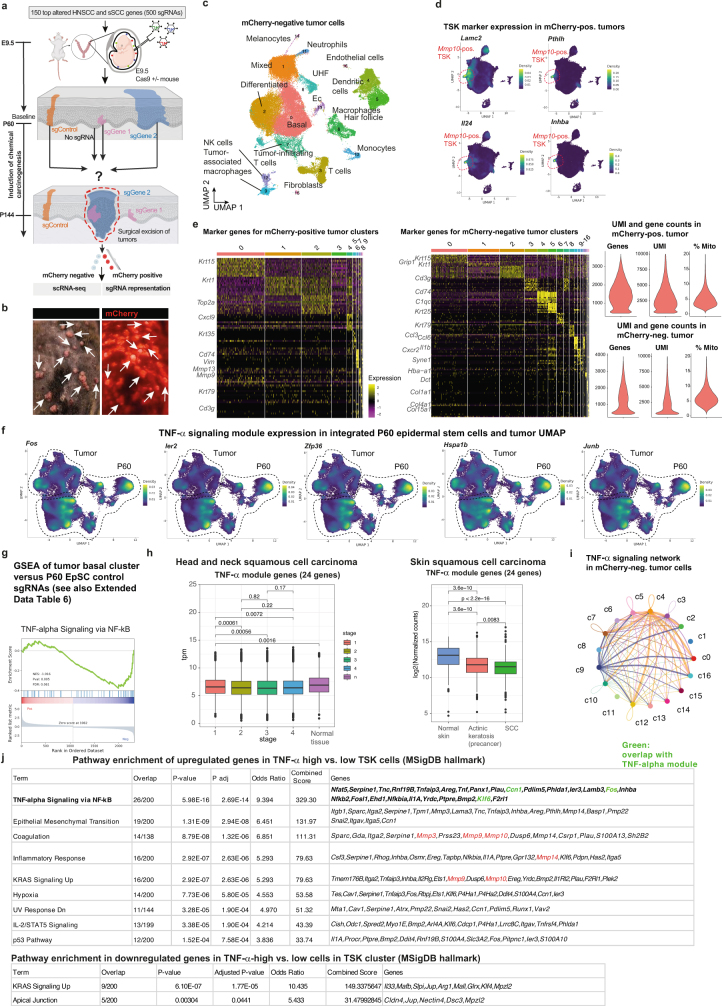
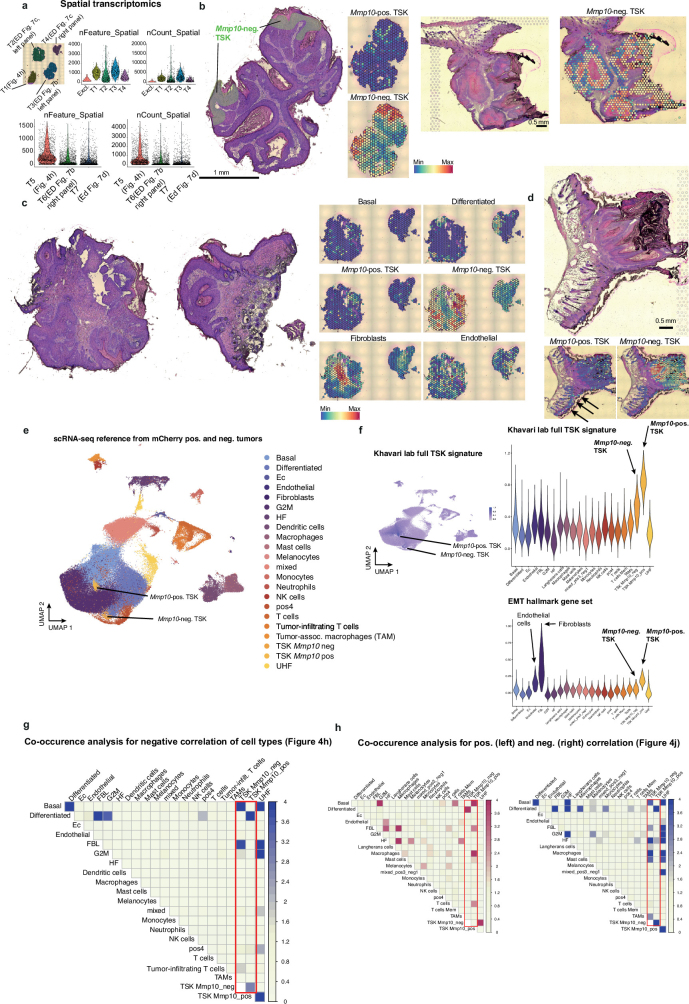
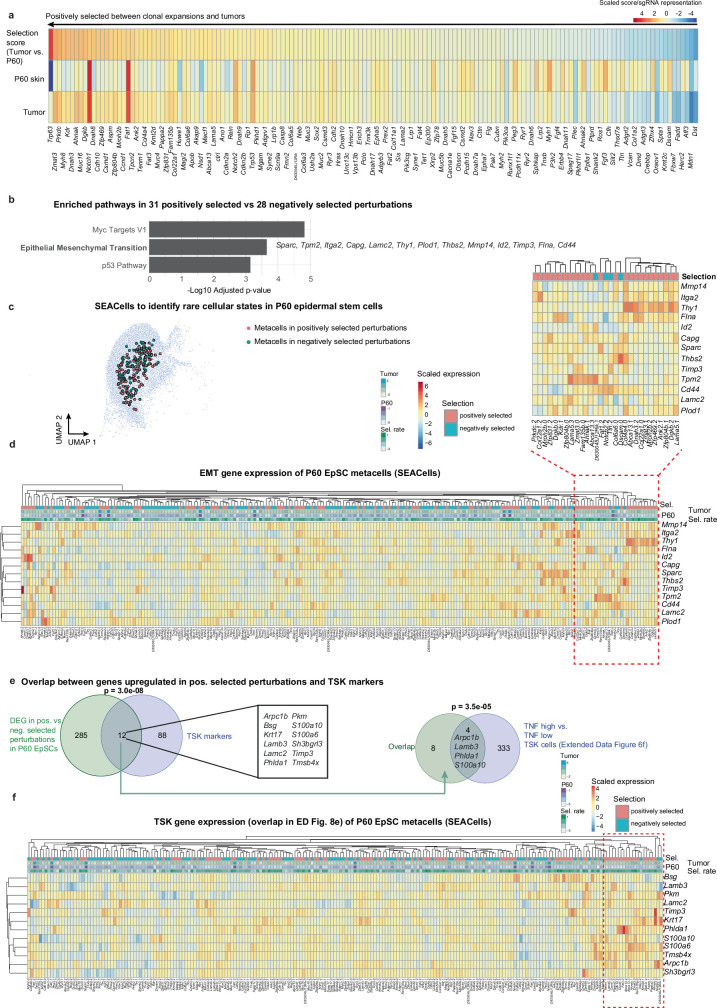
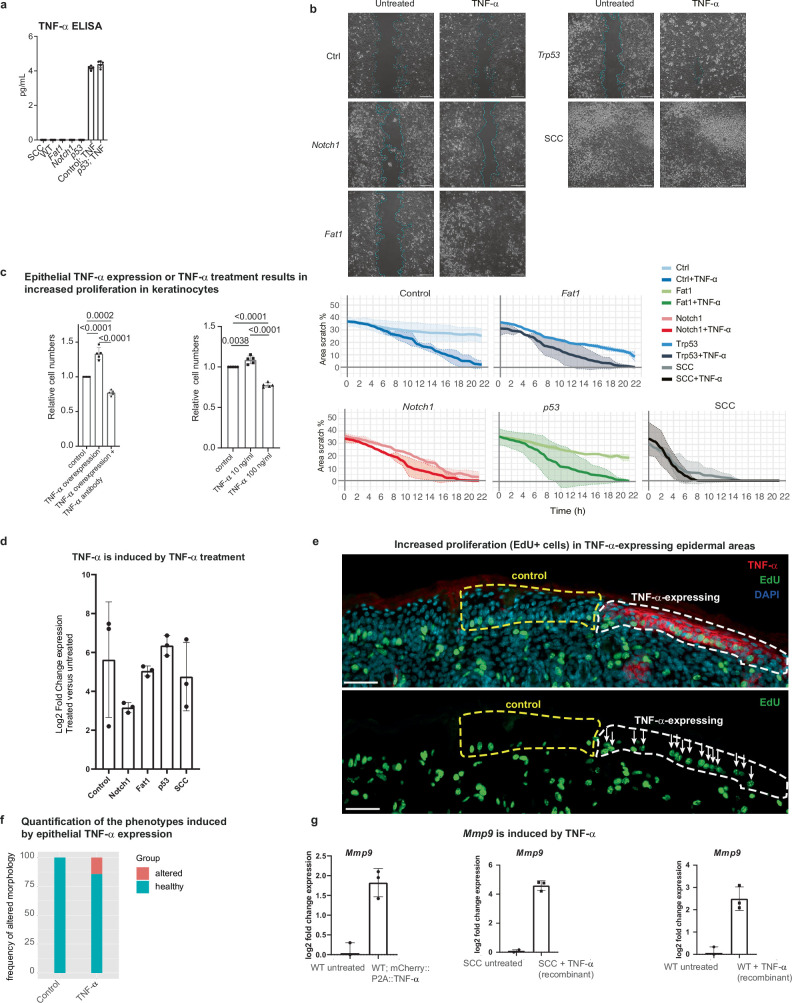
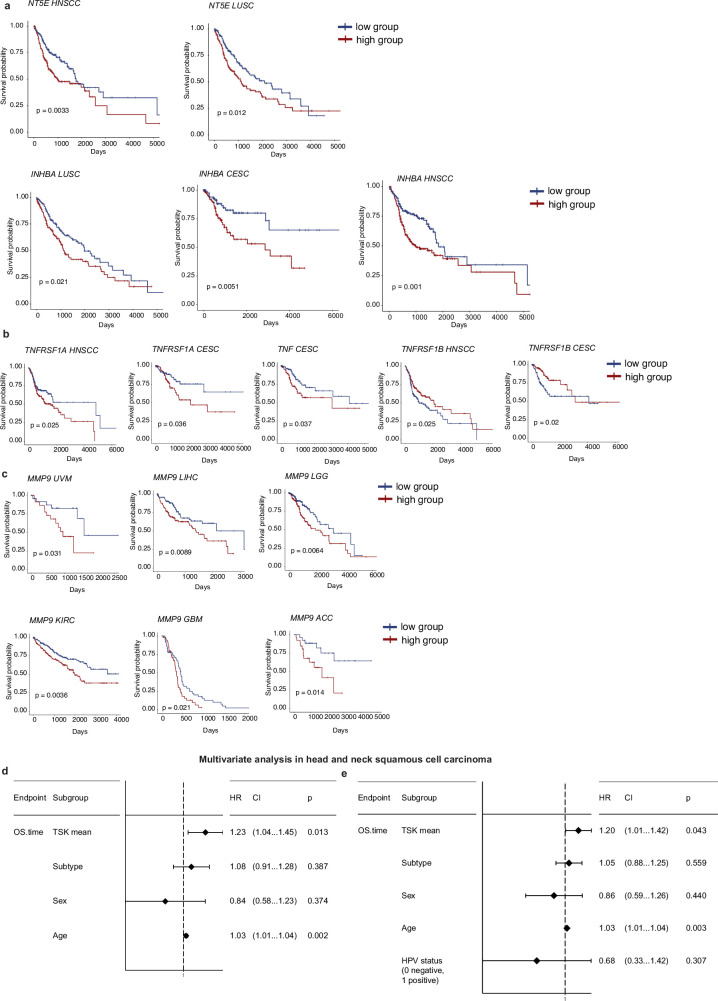
The tumour evolution model posits that malignant transformation is preceded by randomly distributed driver mutations in cancer genes, which cause clonal expansions in phenotypically normal tissues. Although clonal expansions can remodel entire tissues1-3, the mechanisms that result in only a small number of clones transforming into malignant tumours remain unknown. Here we develop an in vivo single-cell CRISPR strategy to systematically investigate tissue-wide clonal dynamics of the 150 most frequently mutated squamous cell carcinoma genes. We couple ultrasound-guided in utero lentiviral microinjections, single-cell RNA sequencing and guide capture to longitudinally monitor clonal expansions and document their underlying gene programmes at single-cell transcriptomic resolution. We uncover a tumour necrosis factor (TNF) signalling module, which is dependent on TNF receptor 1 and involving macrophages, that acts as a generalizable driver of clonal expansions in epithelial tissues. Conversely, during tumorigenesis, the TNF signalling module is downregulated. Instead, we identify a subpopulation of invasive cancer cells that switch to an autocrine TNF gene programme associated with epithelial-mesenchymal transition. Finally, we provide in vivo evidence that the autocrine TNF gene programme is sufficient to mediate invasive properties and show that the TNF signature correlates with shorter overall survival of patients with squamous cell carcinoma. Collectively, our study demonstrates the power of applying in vivo single-cell CRISPR screening to mammalian tissues, unveils distinct TNF programmes in tumour evolution and highlights the importance of understanding the relationship between clonal expansions in epithelia and tumorigenesis.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
