

Identification of plant transcriptional activation domains
- PMID: 39020176
- PMCID: PMC11589624
- DOI: 10.1038/s41586-024-07707-3
Identification of plant transcriptional activation domains
Abstract
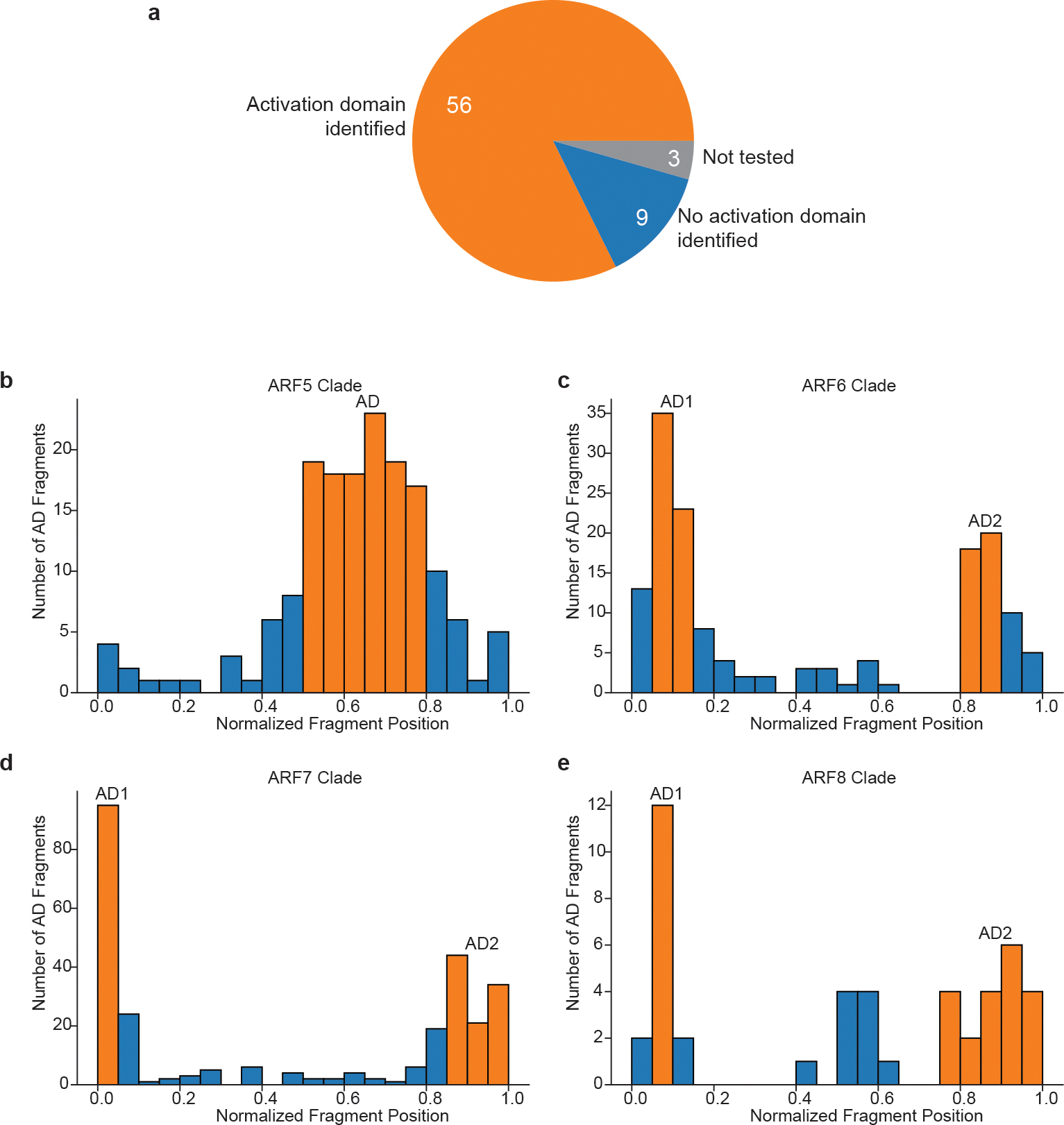
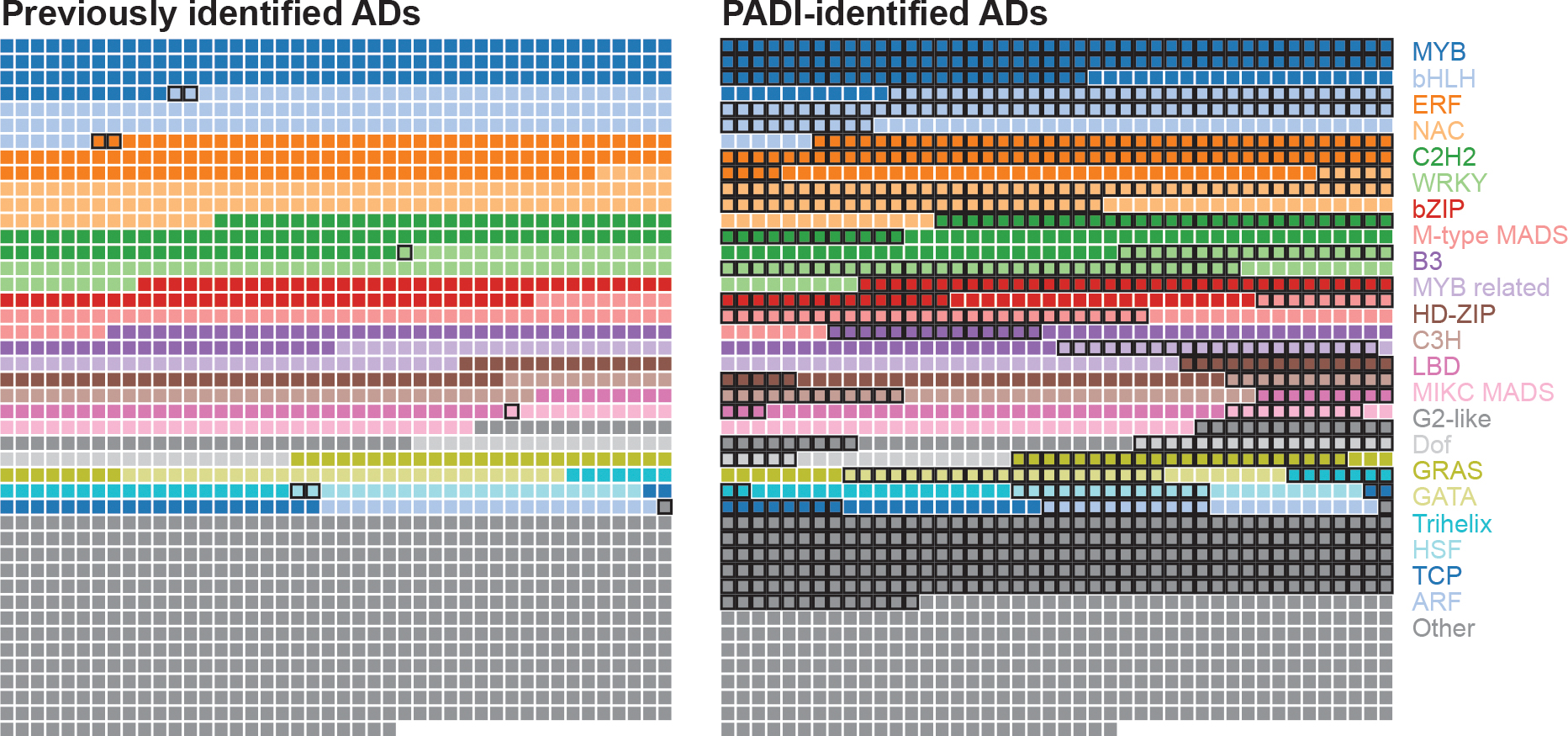
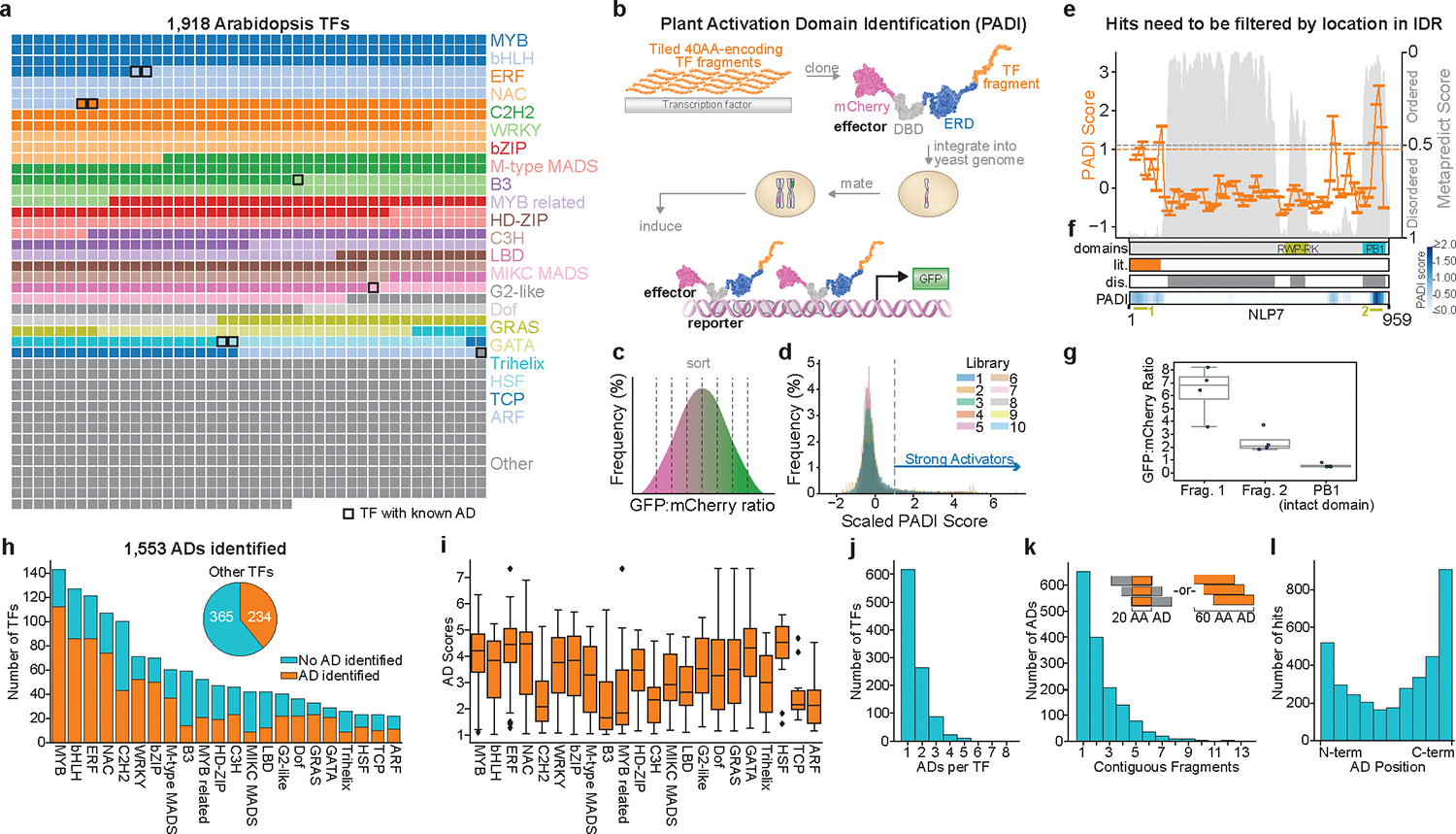
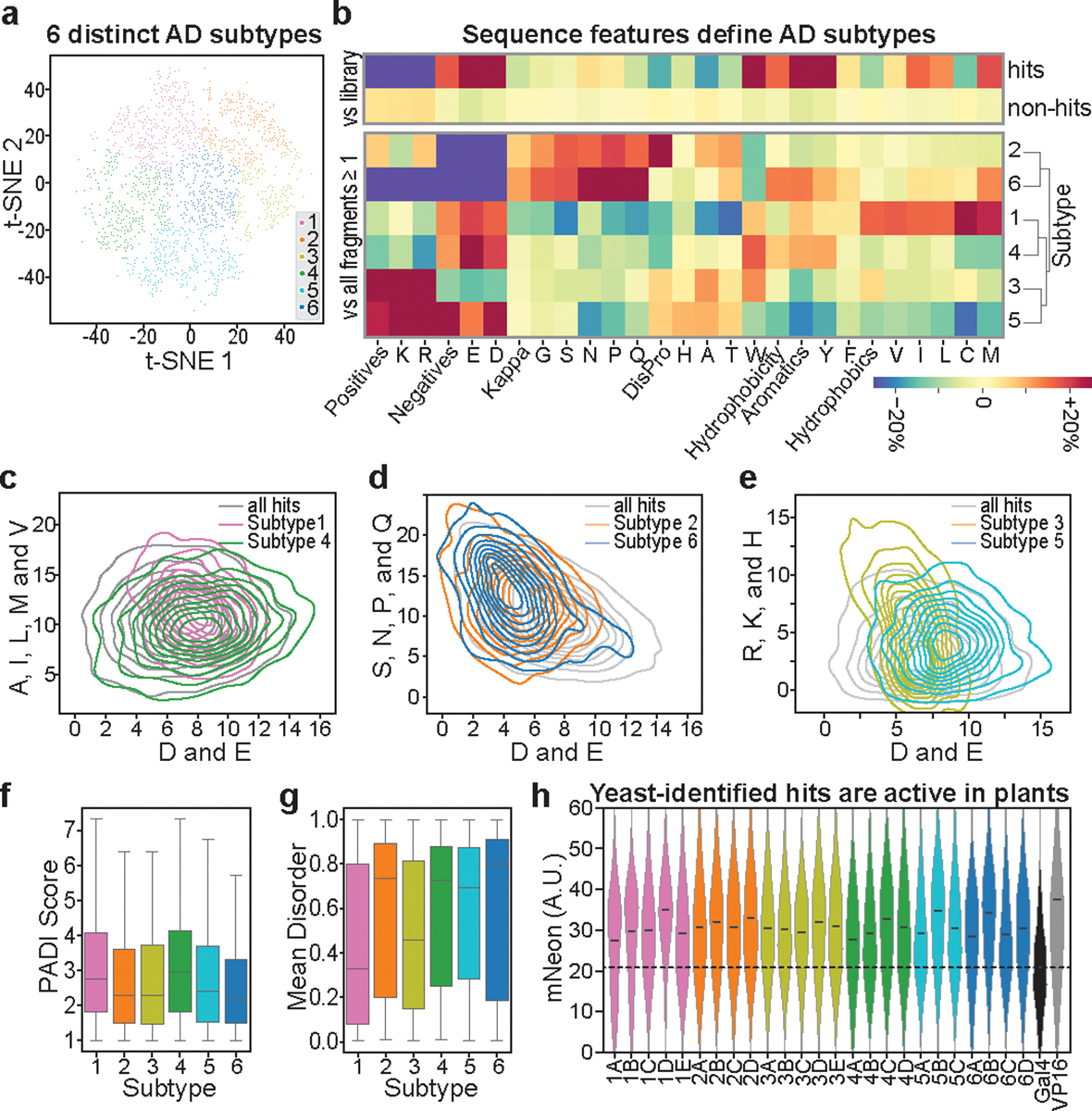
Gene expression in Arabidopsis is regulated by more than 1,900 transcription factors (TFs), which have been identified genome-wide by the presence of well-conserved DNA-binding domains. Activator TFs contain activation domains (ADs) that recruit coactivator complexes; however, for nearly all Arabidopsis TFs, we lack knowledge about the presence, location and transcriptional strength of their ADs1. To address this gap, here we use a yeast library approach to experimentally identify Arabidopsis ADs on a proteome-wide scale, and find that more than half of the Arabidopsis TFs contain an AD. We annotate 1,553 ADs, the vast majority of which are, to our knowledge, previously unknown. Using the dataset generated, we develop a neural network to accurately predict ADs and to identify sequence features that are necessary to recruit coactivator complexes. We uncover six distinct combinations of sequence features that result in activation activity, providing a framework to interrogate the subfunctionalization of ADs. Furthermore, we identify ADs in the ancient AUXIN RESPONSE FACTOR family of TFs, revealing that AD positioning is conserved in distinct clades. Our findings provide a deep resource for understanding transcriptional activation, a framework for examining function in intrinsically disordered regions and a predictive model of ADs.
© 2024. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Competing Interests
LCS is on the science advisory board of Prose Foods. RS is founder of Raleigh Biosciences. All other authors declare no competing interests.
Figures











References
-
- O’Malley RC et al. Cistrome and Epicistrome Features Shape the Regulatory DNA Landscape. Cell 166, 1598 (2016). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
