

The duality of amyloid-β: its role in normal and Alzheimer's disease states
- PMID: 39020435
- PMCID: PMC11256416
- DOI: 10.1186/s13041-024-01118-1
The duality of amyloid-β: its role in normal and Alzheimer's disease states
Abstract
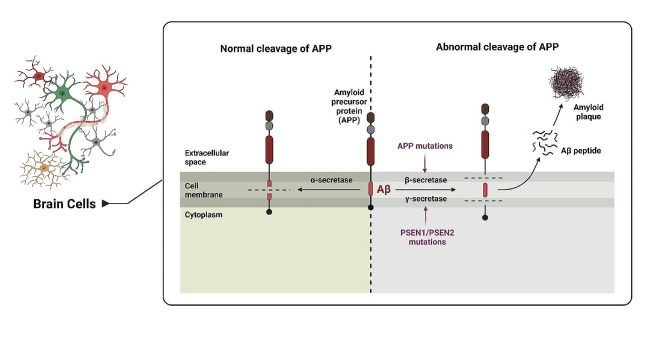
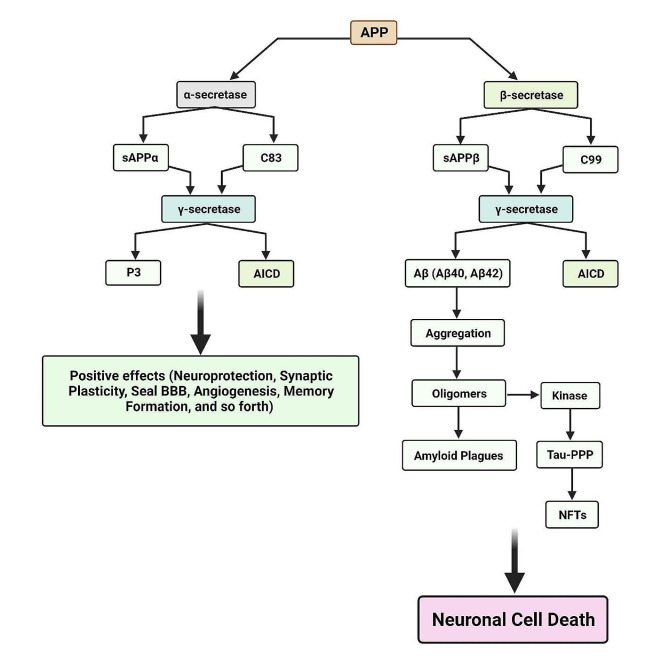
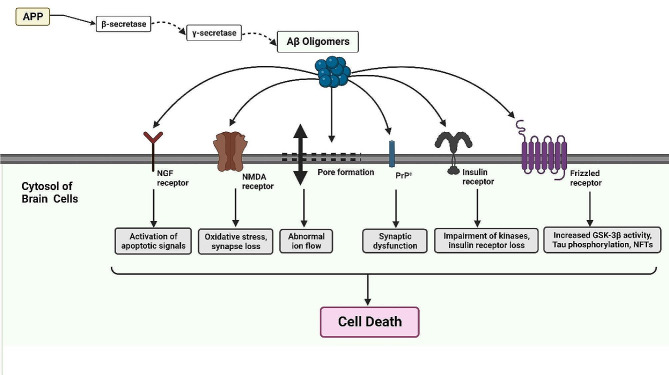
Alzheimer's disease (AD) is a degenerative neurological condition that gradually impairs cognitive abilities, disrupts memory retention, and impedes daily functioning by impacting the cells of the brain. A key characteristic of AD is the accumulation of amyloid-beta (Aβ) plaques, which play pivotal roles in disease progression. These plaques initiate a cascade of events including neuroinflammation, synaptic dysfunction, tau pathology, oxidative stress, impaired protein clearance, mitochondrial dysfunction, and disrupted calcium homeostasis. Aβ accumulation is also closely associated with other hallmark features of AD, underscoring its significance. Aβ is generated through cleavage of the amyloid precursor protein (APP) and plays a dual role depending on its processing pathway. The non-amyloidogenic pathway reduces Aβ production and has neuroprotective and anti-inflammatory effects, whereas the amyloidogenic pathway leads to the production of Aβ peptides, including Aβ40 and Aβ42, which contribute to neurodegeneration and toxic effects in AD. Understanding the multifaceted role of Aβ, particularly in AD, is crucial for developing effective therapeutic strategies that target Aβ metabolism, aggregation, and clearance with the aim of mitigating the detrimental consequences of the disease. This review aims to explore the mechanisms and functions of Aβ under normal and abnormal conditions, particularly in AD, by examining both its beneficial and detrimental effects.
Keywords: Alzheimer’s disease; Beta amyloid; Cognitive decline; Long-term potentiation; Neuroinflammation; Neuroprotection; Neurotoxicity.
© 2024. The Author(s).
Conflict of interest statement
There are no conflicting interests to be stated.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
