

Combined clinical, structural and cellular studies discriminate pathogenic and benign TRPV4 variants
- PMID: 39021275
- PMCID: PMC12054728
- DOI: 10.1093/brain/awae243
Combined clinical, structural and cellular studies discriminate pathogenic and benign TRPV4 variants
Abstract
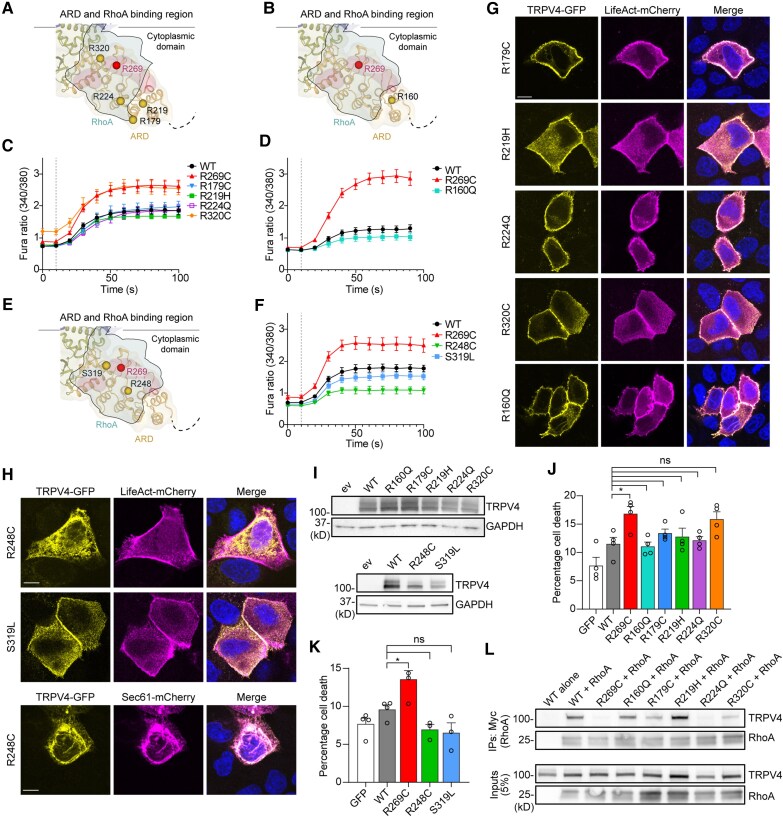
Dominant mutations in the calcium-permeable ion channel TRPV4 (transient receptor potential vanilloid 4) cause diverse and largely distinct channelopathies, including inherited forms of neuromuscular disease, skeletal dysplasias and arthropathy. Pathogenic TRPV4 mutations cause gain of ion channel function and toxicity that can be rescued by small molecule TRPV4 antagonists in cellular and animal models, suggesting that TRPV4 antagonism could be therapeutic for patients. Numerous variants in TRPV4 have been detected with targeted and whole exome/genome sequencing, but for the vast majority, their pathogenicity remains unclear. Here, we used a combination of clinical information and experimental structure-function analyses to evaluate 30 TRPV4 variants across various functional protein domains. We report clinical features of seven patients with TRPV4 variants of unknown significance and provide extensive functional characterization of these and an additional 17 variants, including structural position, ion channel function, subcellular localization, expression level, cytotoxicity and protein-protein interactions. We find that gain-of-function mutations within the TRPV4 intracellular ankyrin repeat domain target charged amino acid residues important for RhoA interaction, whereas ankyrin repeat domain residues outside of the RhoA interface have normal or reduced ion channel activity. We further identify a cluster of gain-of-function variants within the intracellular intrinsically disordered region that may cause toxicity via altered interactions with membrane lipids. In contrast, assessed variants in the transmembrane domain and other regions of the intrinsically disordered region do not cause gain of function and are likely benign. Clinical features associated with gain of function and cytotoxicity include congenital onset of disease, vocal cord weakness and motor-predominant disease, whereas patients with likely benign variants often demonstrated late-onset and sensory-predominant disease. These results provide a framework for assessing additional TRPV4 variants with respect to likely pathogenicity, which will yield critical information to inform patient selection for future clinical trials for TRPV4 channelopathies.
Keywords: Charcot-Marie-Tooth disease; RhoA; TRPV4; calcium signalling; neuropathy; skeletal dysplasia.
© The Author(s) 2024. Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For commercial re-use, please contact reprints@oup.com for reprints and translation rights for reprints. All other permissions can be obtained through our RightsLink service via the Permissions link on the article page on our site—for further information please contact journals.permissions@oup.com.
Conflict of interest statement
M.H., S.T., T.O.C., C.J.S and B.A.M. are participating in an investigator-initiated natural history study for TRPV4 neuropathy (NCT05600764) that is supported by Actio Biosciences. C.J.S. is supported by Actio Biosciences for laboratory-based studies of TRPV4 channelopathy.
Figures





References
-
- McCray BA, Schindler A, Hoover-Fong JE, Sumner CJ. Autosomal Dominant TRPV4 Disorders. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds. GeneReviews®. University of Washington; 2014. - PubMed
-
- White JP, Cibelli M, Urban L, Nilius B, McGeown JG, Nagy I. TRPV4: Molecular conductor of a diverse orchestra. Physiol Rev. 2016;96:911–973. - PubMed
-
- Echaniz-Laguna A, Dubourg O, Carlier P, et al. Phenotypic spectrum and incidence of TRPV4 mutations in patients with inherited axonal neuropathy. Neurology. 2014;82:1919–1926. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
