

Amino acid deprivation in cancer cells with compensatory autophagy induction increases sensitivity to autophagy inhibitors
- PMID: 39021618
- PMCID: PMC11253891
- DOI: 10.1080/23723556.2024.2377404
Amino acid deprivation in cancer cells with compensatory autophagy induction increases sensitivity to autophagy inhibitors
Abstract
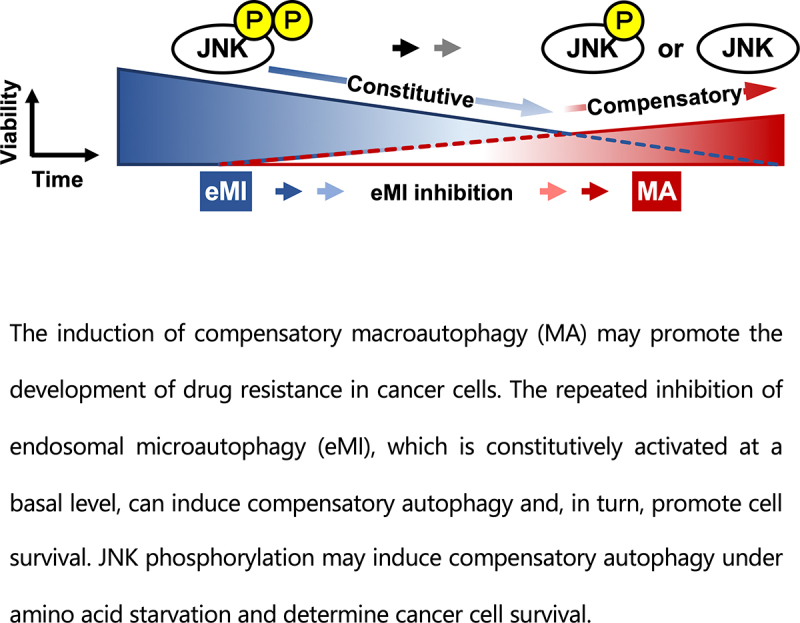
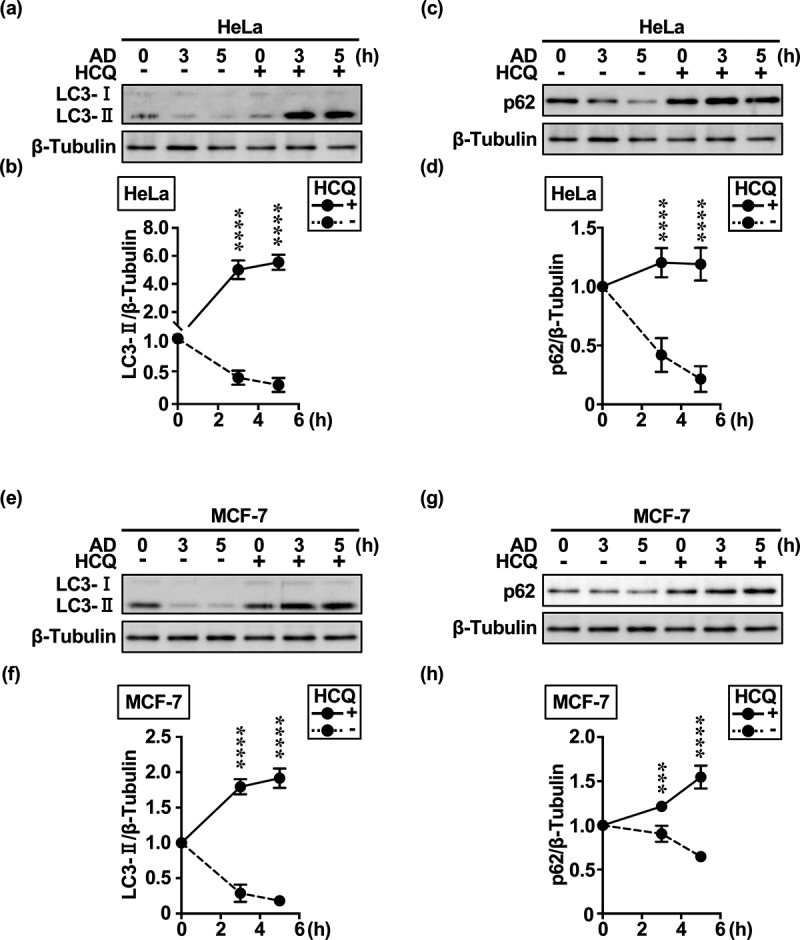
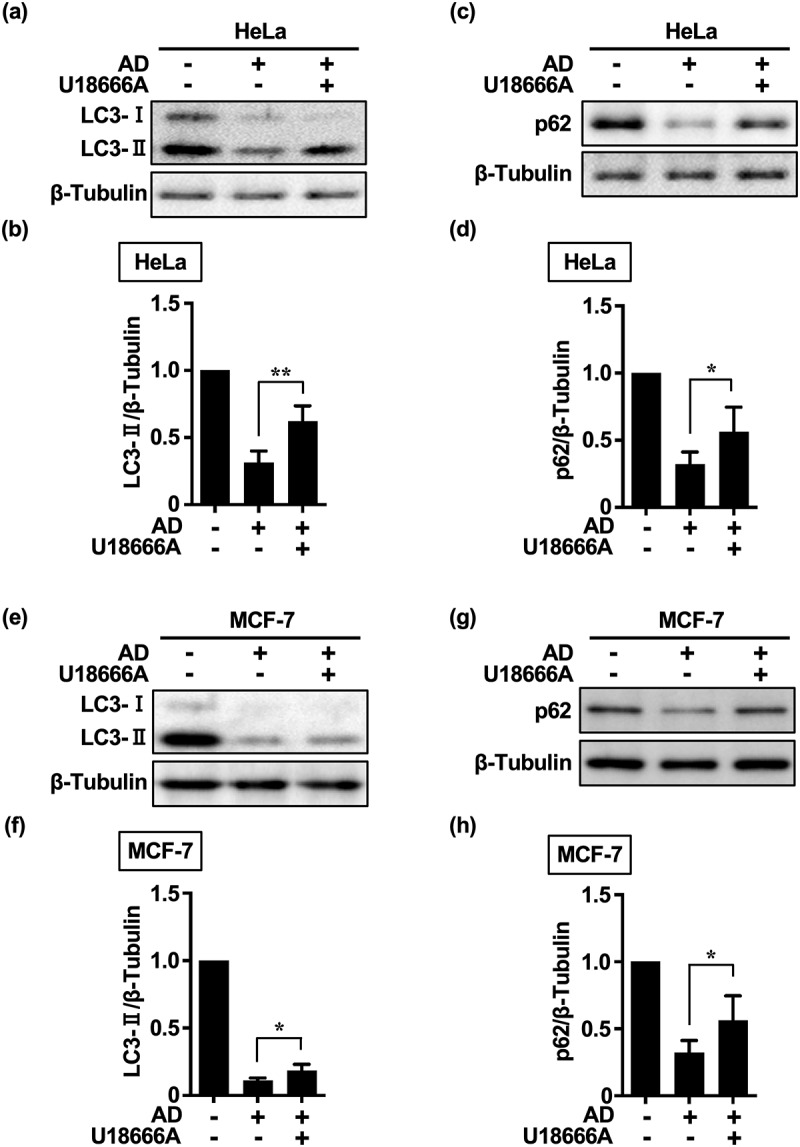
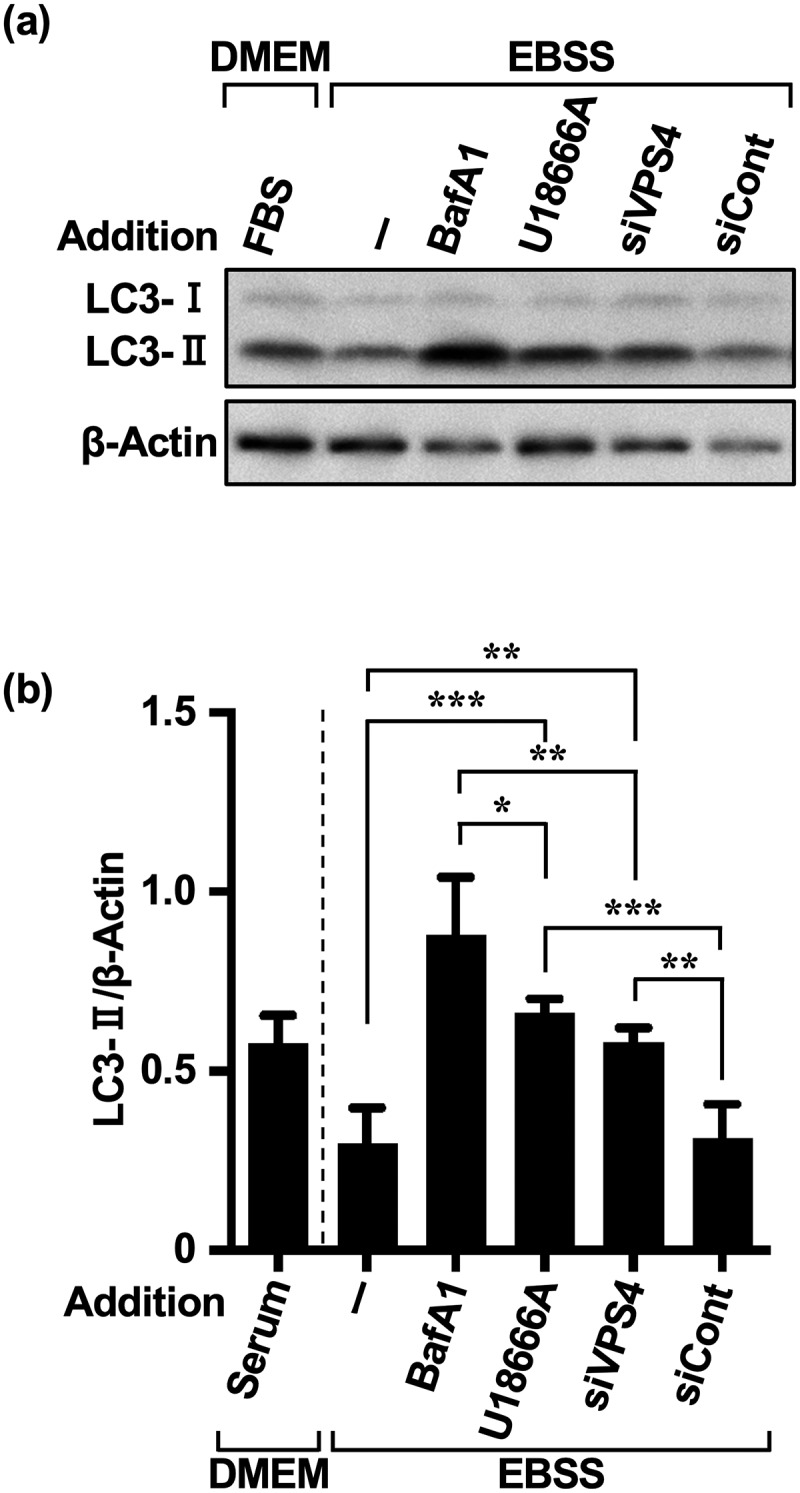
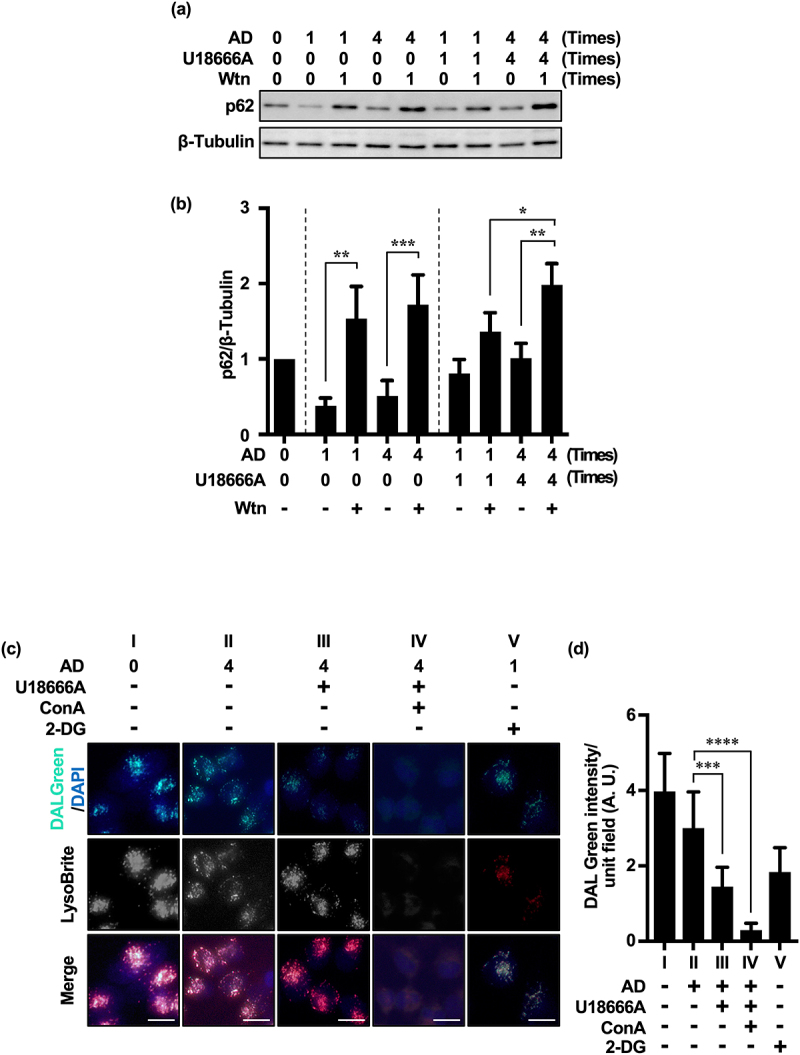
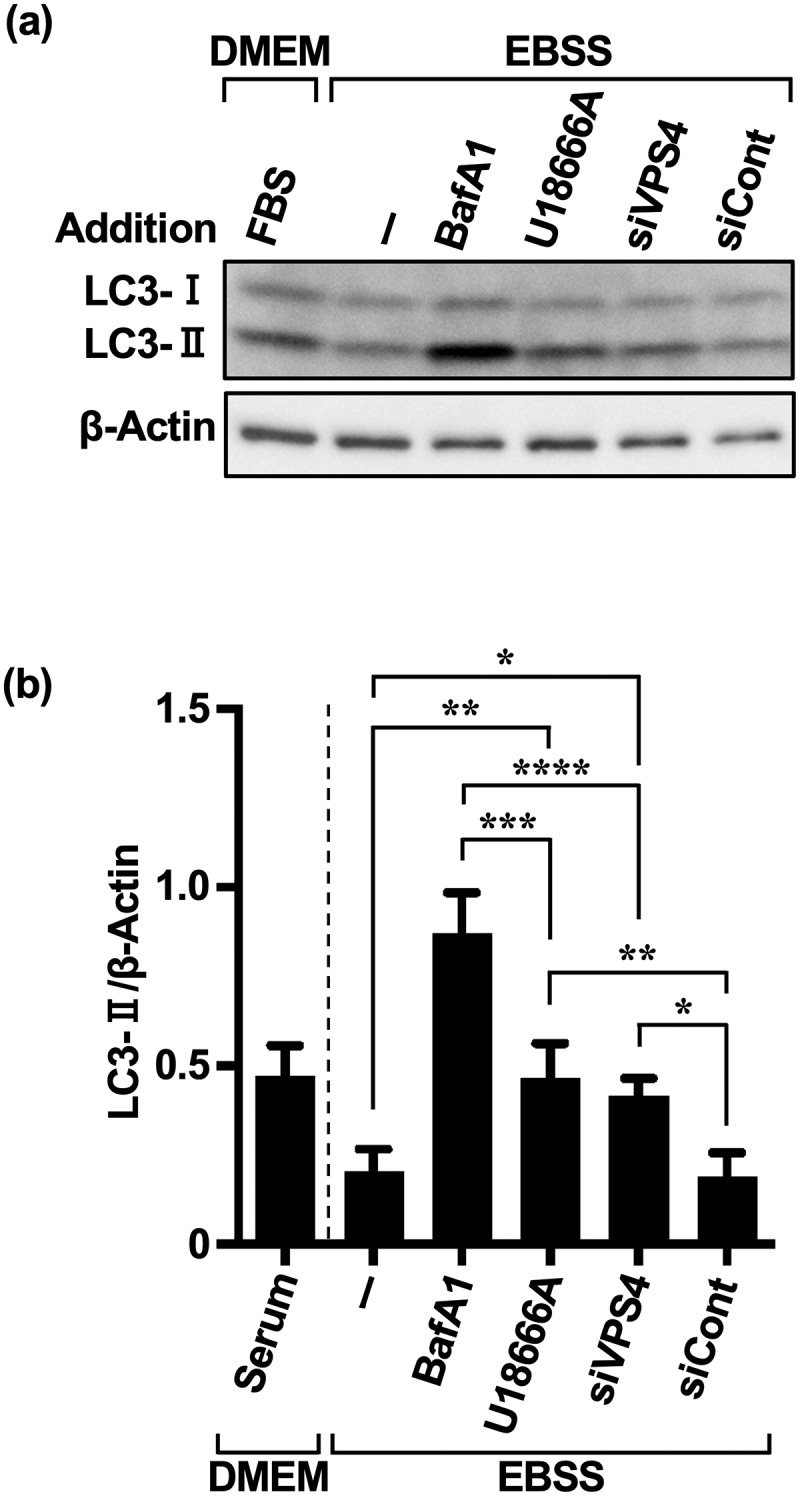
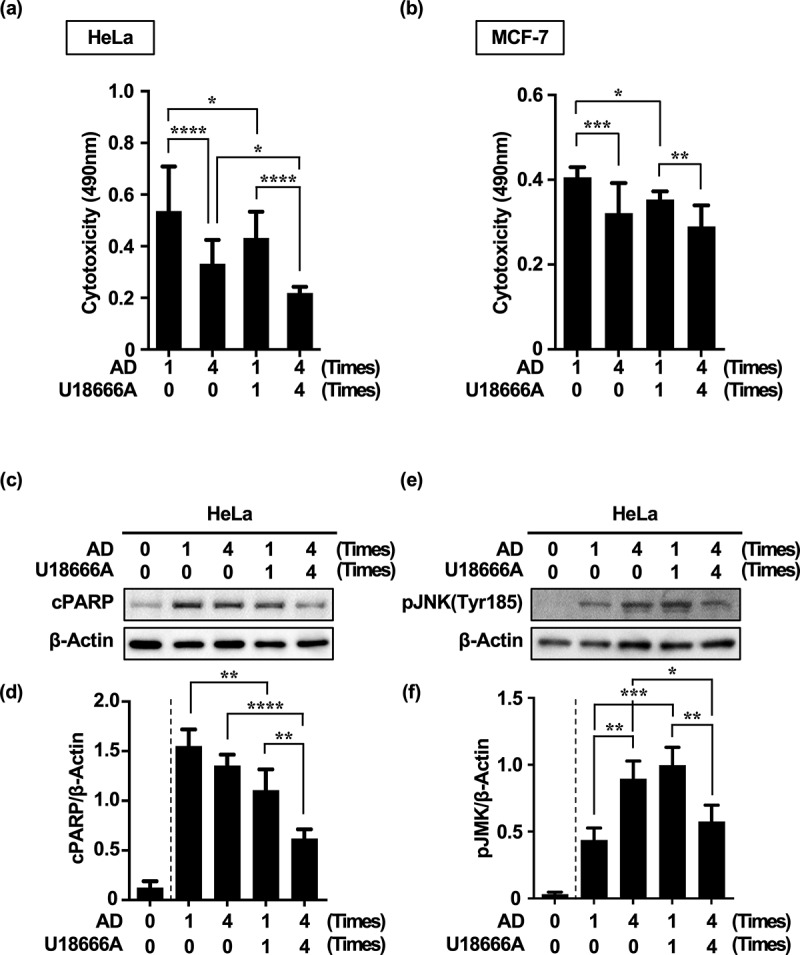
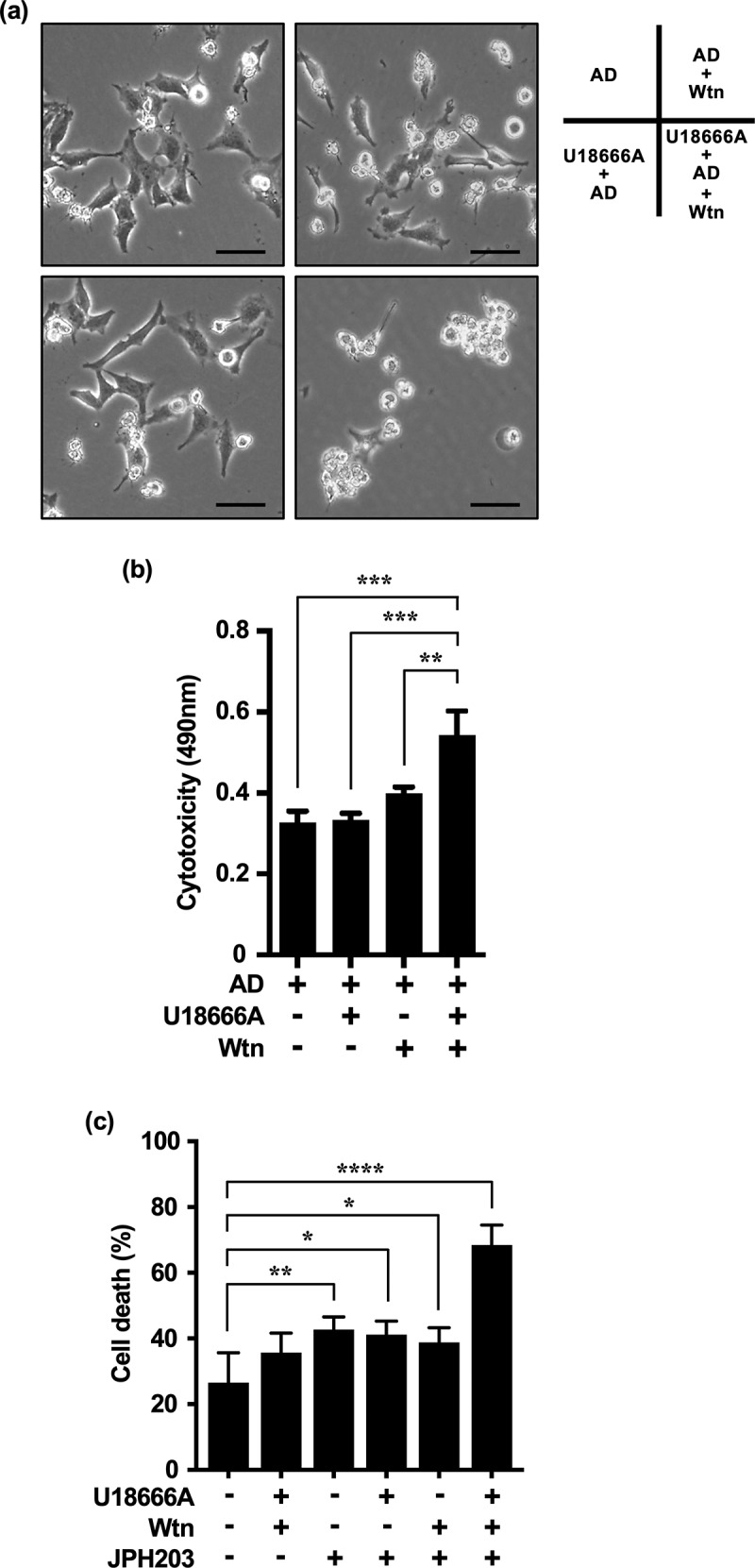
Inhibition of autophagy is an important strategy in cancer therapy. However, prolonged inhibition of certain autophagies in established cancer cells may increase therapeutic resistance, though the underlying mechanisms of its induction and enhancement remain unclear. This study sought to elucidate the mechanisms of therapeutic resistance through repeated autophagy inhibition and amino acid deprivation (AD) in an in vitro model of in vivo chronic nutrient deprivation associated with cancer cell treatment. In the human cervical cancer cell line HeLa and human breast cancer cell line MCF-7, initial extracellular AD induced the immediate expression of endosomal microautophagy (eMI). However, repeated inhibition of eMI with U18666A and extracellular AD induced macroautophagy (MA) to compensate for reduced eMI, simultaneously decreasing cytotoxicity. Here, hyperphosphorylated JNK was transformed into a hypophosphorylated state, suggesting conversion of the cell death signal to a survival signal. In a nutrient medium, cell death could not be induced by MA inhibition. However, since LAT1 inhibitors induce intracellular AD, combining them with MA and eMI inhibitors successfully promoted cell death in resistant cells. Our study identified a novel therapeuic approach for promoting cell death and addressing therapeutic resistance in cancers under autophagy-inhibitor treatment.
Keywords: Amino acid deprivation; amino acid transporter; autophagy; cancer; chemoresistance.
© 2024 The Author(s). Published with license by Taylor & Francis Group, LLC.
Conflict of interest statement
No potential conflict of interest was reported by the author(s).
Figures
Similar articles
-
Differential activation of eMI by distinct forms of cellular stress.Autophagy. 2021 Aug;17(8):1828-1840. doi: 10.1080/15548627.2020.1783833. Epub 2020 Jul 9. Autophagy. 2021. PMID: 32559125 Free PMC article.
-
Selective endosomal microautophagy is starvation-inducible in Drosophila.Autophagy. 2016 Nov;12(11):1984-1999. doi: 10.1080/15548627.2016.1208887. Epub 2016 Aug 3. Autophagy. 2016. PMID: 27487474 Free PMC article.
-
An autophagy inhibitor enhances the inhibition of cell proliferation induced by a proteasome inhibitor in MCF-7 cells.Mol Med Rep. 2012 Jan;5(1):84-8. doi: 10.3892/mmr.2011.590. Epub 2011 Sep 19. Mol Med Rep. 2012. PMID: 21931937
-
Diving into the Evolutionary History of HSC70-Linked Selective Autophagy Pathways: Endosomal Microautophagy and Chaperone-Mediated Autophagy.Cells. 2022 Jun 16;11(12):1945. doi: 10.3390/cells11121945. Cells. 2022. PMID: 35741074 Free PMC article. Review.
-
Cell death and autophagy: cytokines, drugs, and nutritional factors.Toxicology. 2008 Dec 30;254(3):147-57. doi: 10.1016/j.tox.2008.07.048. Epub 2008 Jul 23. Toxicology. 2008. PMID: 18694801 Review.
References
LinkOut - more resources
Full Text Sources
Research Materials