

[Influence of Clinical Practice Guidelines on the Diagnosis and Treatment of Idiopathic Pulmonary Fibrosis. Data from the Registry of the Spanish Society of Pulmonology and Thoracic Surgery]
- PMID: 39021619
- PMCID: PMC11253673
- DOI: 10.1016/j.opresp.2024.100334
[Influence of Clinical Practice Guidelines on the Diagnosis and Treatment of Idiopathic Pulmonary Fibrosis. Data from the Registry of the Spanish Society of Pulmonology and Thoracic Surgery]
Abstract
Objective: The objective of the study was to analyze the diagnostic process and the time until the start of treatment of patients with idiopathic pulmonary fibrosis in relation to the publication of successive clinical practice guide.
Material and methods: Multicenter, observational, ambispective study, in which patients includes in the idiopathic pulmonary fibrosis registry of the Spanish Society of Pulmonologist and Thoracic Surgery were analyzed. An electronic data collection notebook was enabled on the society's website. Sociodemographic and clinical variables were collected at diagnosis and follow-up of the patients.
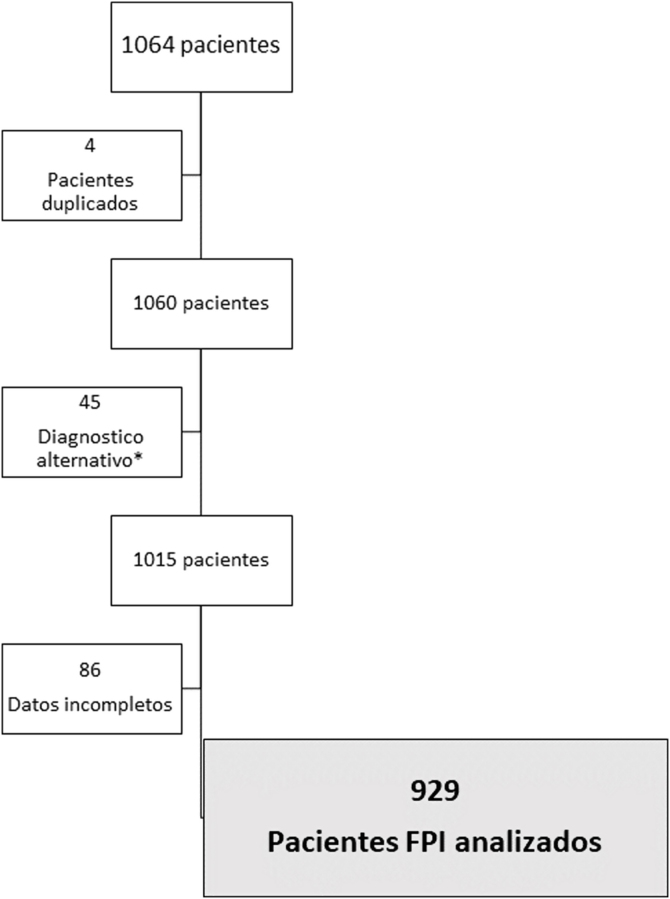
Results: From January 2012 to december 2019, 1064 patients were included in the registry, with 929 finally analyzed. The diagnosis process varied depending on the year in which it was performed, and the radiological pattern observed in the high-resolution computed tomography. Up to 26.3% of the cases (244) were diagnosed with chest high-resolution computed tomography and clinical evaluation. Surgical biopsy was used up to 50.2% of cases diagnosed before 2011, while it has been used in 14.2% since 2018. The median time from the onset of symptoms to diagnosis was 360 days (IQR 120-720), taking more than 2 years in the 21.0% of patients. A percentage of 79.4 of patients received antifibrotic treatment. The average time from diagnosis to the antifibrotic treatment has been 309 ± 596.5 days, with a median of 49 (IQR 0-307).
Conclusions: The diagnostic process, including the time until diagnosis and the type of test used, has changed from 2011 to 2019, probably due to advances in clinical research and the publication of diagnostic-therapeutic consensus guidelines.
Objetivo: El objetivo de este estudio fue analizar el proceso diagnóstico de los pacientes con fibrosis pulmonar idiopática en España, desde el inicio de los síntomas hasta el diagnóstico y tratamiento antifibrótico, en relación con la publicación de las sucesivas guías de práctica clínica.
Material y métodos: Estudio multicéntrico, observacional, ambispectivo, en el que se analizaron los pacientes incluidos en el registro de la fibrosis pulmonar idiopática de la Sociedad Española de Neumología y Cirugía Torácica. Para ello se habilitó un cuaderno electrónico de recogida de datos en la web de la sociedad. Se recogieron variables sociodemográficas y clínicas al diagnóstico y seguimiento de los pacientes.
Resultados: Desde enero de 2012 hasta diciembre de 2019 se incluyeron 1.064 pacientes, siendo finalmente analizados 929. El proceso diagnóstico varió en función del año en el que se realizó el diagnóstico y el patrón radiológico observado en la tomografía computarizada de alta resolución. En 244 (26,3%) pacientes, el diagnóstico se realizó con tomografía computarizada de alta resolución de tórax y evaluación clínica. La biopsia quirúrgica se utilizó hasta en el 50,2% de los casos diagnosticados antes del 2011, y en un 14,2% a partir de 2018. La mediana de tiempo que transcurre desde el inicio de los síntomas hasta el diagnóstico es de 360 días (RIC 120-720), siendo mayor de 2 años en el 21,0% de los pacientes. Recibieron tratamiento antifibrótico al 79,4% de los pacientes. El tiempo desde el diagnóstico hasta el inicio del tratamiento fue de 309 ± 596,5 días, con una mediana de 49 (RIC 0-307).
Conclusiones: El proceso diagnóstico, incluyendo el tiempo hasta el diagnóstico y el tipo de pruebas utilizadas, ha ido cambiando desde 2011 hasta 2019, probablemente debido el avance en la investigación clínica y la publicación de guías consenso diagnóstico-terapéuticas.
Keywords: Idiopathic pulmonary fibrosis; Interstitial lung disease; Multidisciplinary diagnosis team.
© 2024 Sociedad Española de Neumología y Cirugía Torácica (SEPAR). Published by Elsevier España, S.L.U.
Figures
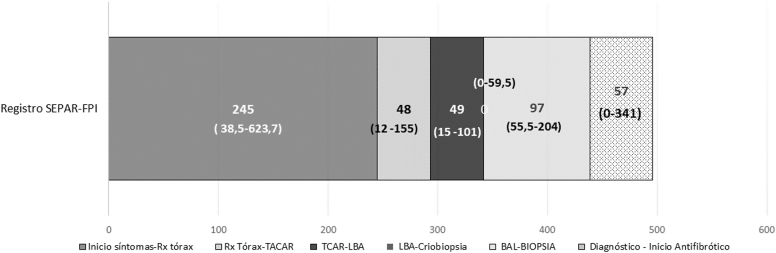
 LBA: lavado broncoalveolar; Rx tórax: radiografía de tórax; TACAR: tomografía axial computarizada de alta resolución.
LBA: lavado broncoalveolar; Rx tórax: radiografía de tórax; TACAR: tomografía axial computarizada de alta resolución.Similar articles
-
Deep learning for classifying fibrotic lung disease on high-resolution computed tomography: a case-cohort study.Lancet Respir Med. 2018 Nov;6(11):837-845. doi: 10.1016/S2213-2600(18)30286-8. Epub 2018 Sep 16. Lancet Respir Med. 2018. PMID: 30232049
-
Russian Registry of Idiopathic Pulmonary Fibrosis: Clinical Features, Treatment Management, and Outcomes.Life (Basel). 2023 Feb 3;13(2):435. doi: 10.3390/life13020435. Life (Basel). 2023. PMID: 36836792 Free PMC article.
-
[Standard technical specifications for methacholine chloride (Methacholine) bronchial challenge test (2023)].Zhonghua Jie He He Hu Xi Za Zhi. 2024 Feb 12;47(2):101-119. doi: 10.3760/cma.j.cn112147-20231019-00247. Zhonghua Jie He He Hu Xi Za Zhi. 2024. PMID: 38309959 Chinese.
-
[Diffuse idiopathic interstitial pneumonias. International multidisciplinary consensus classification by the American Thoracic Society and the European Respiratory Society, principal clinico-pathological entities, and diagnosis].Rev Mal Respir. 2004 Apr;21(2 Pt 1):299-318. doi: 10.1016/s0761-8425(04)71288-7. Rev Mal Respir. 2004. PMID: 15211238 Review. French.
-
French practical guidelines for the diagnosis and management of idiopathic pulmonary fibrosis - 2021 update. Full-length version.Respir Med Res. 2023 Jun;83:100948. doi: 10.1016/j.resmer.2022.100948. Epub 2022 Aug 4. Respir Med Res. 2023. PMID: 36630775 Review.
References
-
- Xaubet A., Ancochea J., Morell F., Rodriguez-Arias J.M., Villena V., Blanquer R., et al. Spanish Group on Interstitial Lung Diseases, SEPAR Report on the incidence of interstitial lung diseases in Spain. Sarcoidosis Vasc Diffuse Lung Dis. 2004;21:64–70. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
