

Targeting Cholesterol Biosynthesis with Statins Synergizes with AKT Inhibitors in Triple-Negative Breast Cancer
- PMID: 39024548
- PMCID: PMC11443248
- DOI: 10.1158/0008-5472.CAN-24-0970
Targeting Cholesterol Biosynthesis with Statins Synergizes with AKT Inhibitors in Triple-Negative Breast Cancer
Abstract
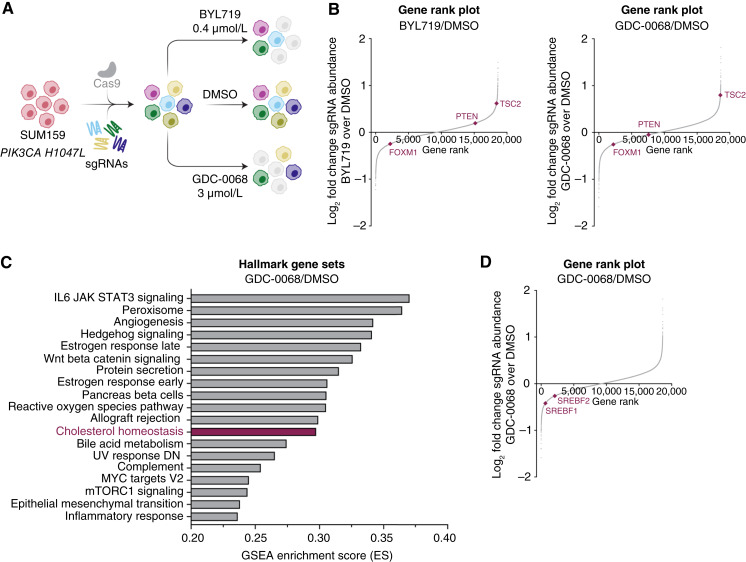
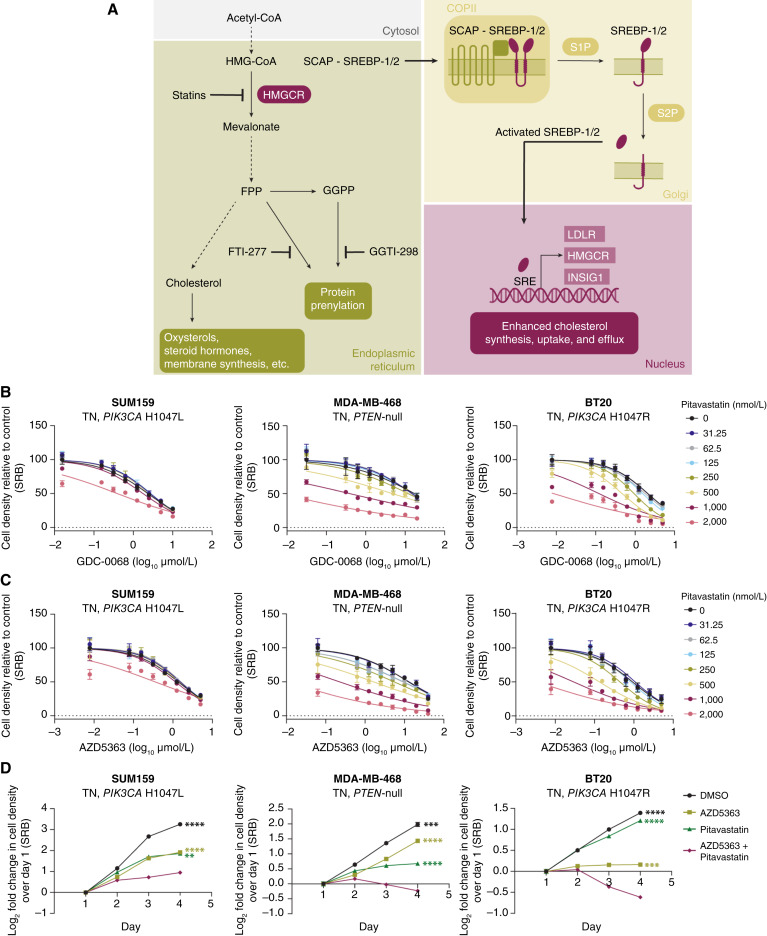
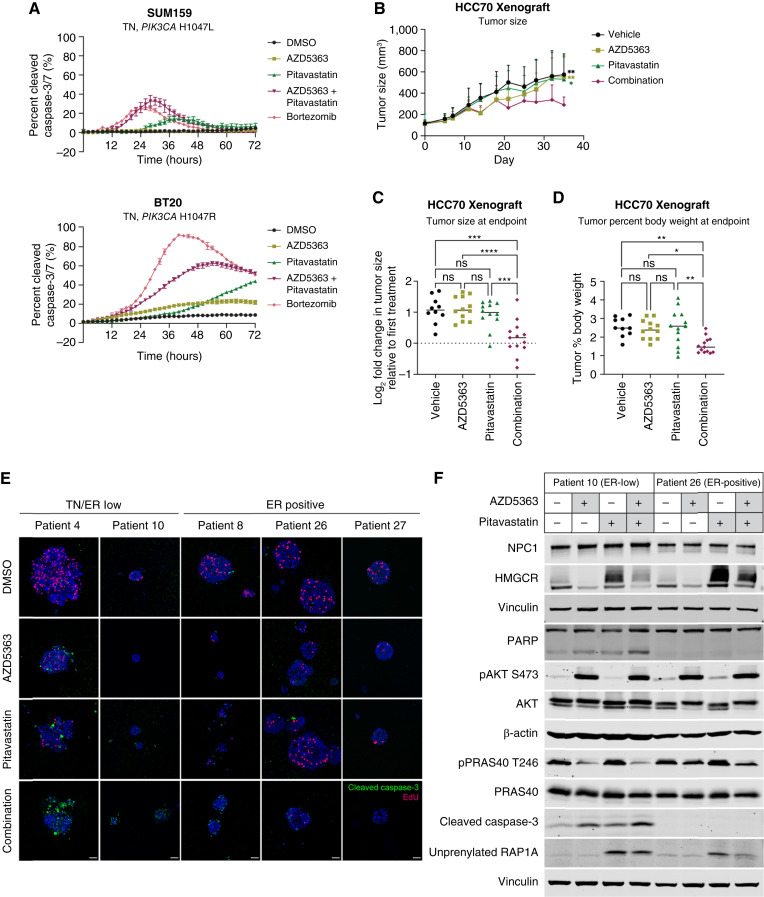
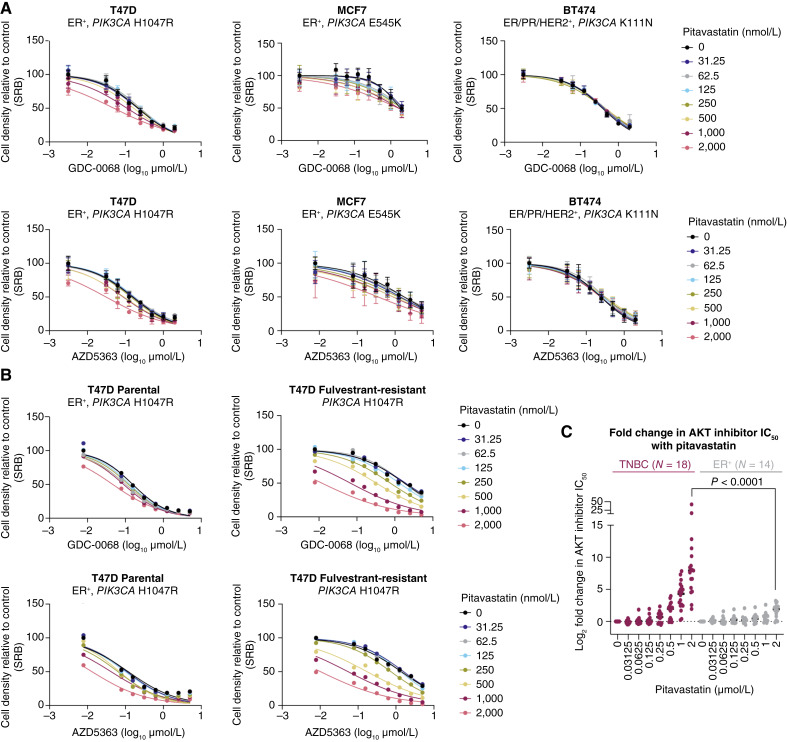
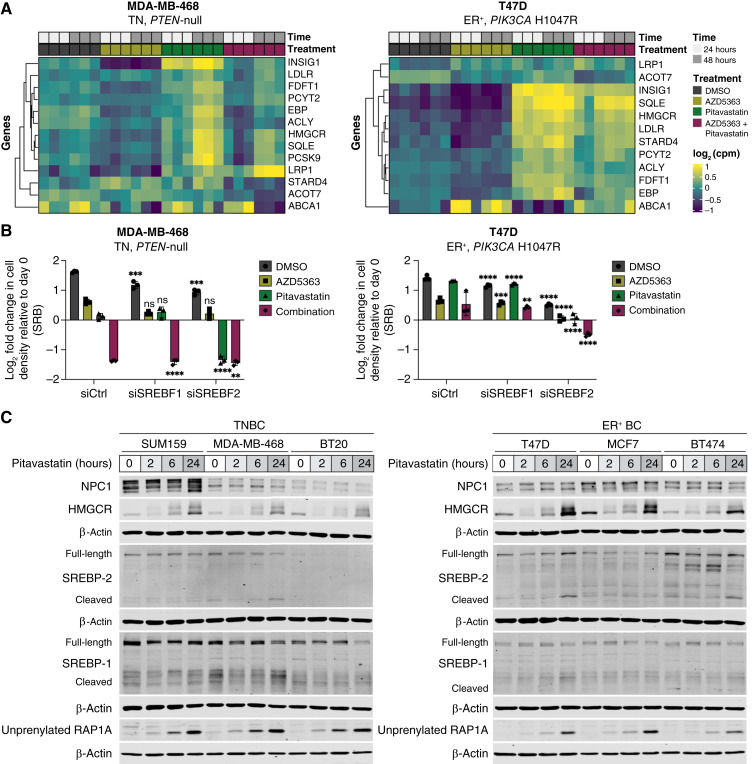
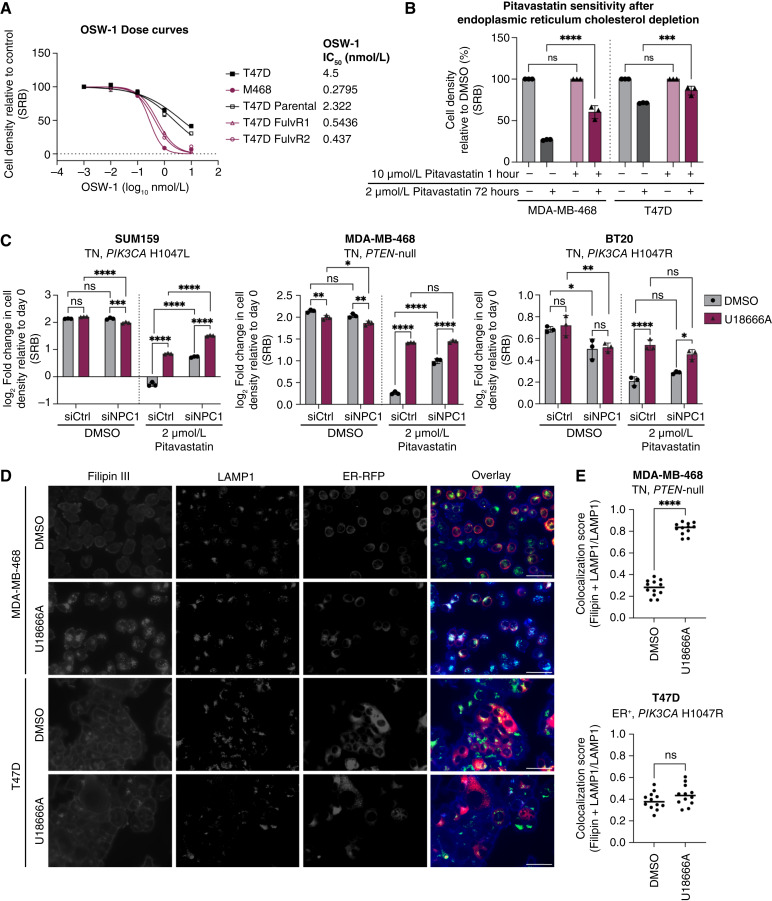
Triple-negative breast cancer (TNBC) is responsible for a disproportionate number of breast cancer patient deaths due to extensive molecular heterogeneity, high recurrence rates, and lack of targeted therapies. Dysregulation of the phosphoinositide 3-kinase (PI3K)/AKT pathway occurs in approximately 50% of TNBC patients. Here, we performed a genome-wide CRISPR/Cas9 screen with PI3Kα and AKT inhibitors to find targetable synthetic lethalities in TNBC. Cholesterol homeostasis was identified as a collateral vulnerability with AKT inhibition. Disruption of cholesterol homeostasis with pitavastatin synergized with AKT inhibition to induce TNBC cytotoxicity in vitro in mouse TNBC xenografts and in patient-derived estrogen receptor (ER)-negative breast cancer organoids. Neither ER-positive breast cancer cell lines nor ER-positive organoids were sensitive to combined AKT inhibitor and pitavastatin. Mechanistically, TNBC cells showed impaired sterol regulatory element-binding protein 2 (SREBP-2) activation in response to single-agent or combination treatment with AKT inhibitor and pitavastatin, which was rescued by inhibition of the cholesterol-trafficking protein Niemann-Pick C1 (NPC1). NPC1 loss caused lysosomal cholesterol accumulation, decreased endoplasmic reticulum cholesterol levels, and promoted SREBP-2 activation. Taken together, these data identify a TNBC-specific vulnerability to the combination of AKT inhibitors and pitavastatin mediated by dysregulated cholesterol trafficking. These findings support combining AKT inhibitors with pitavastatin as a therapeutic modality in TNBC. Significance: Two FDA-approved compounds, AKT inhibitors and pitavastatin, synergize to induce cell death in triple-negative breast cancer, motivating evaluation of the efficacy of this combination in clinical trials.
©2024 The Authors; Published by the American Association for Cancer Research.
Conflict of interest statement
J. Högström reports grants from the American Association for Cancer Research, Maud Kuistila Memorial Foundation, Sigrid Jusélius Foundation and Orion Research Foundation during the conduct of the study. D.E. Root reports grants from AbbVie, Bristol Myers Squibb, Janssen, Merck and Vir Biotechnology outside the submitted work. M. Brown reports grants from the Ludwig Center at Harvard and Breast Cancer Research Foundation during the conduct of the study; as well as grants from Novartis and personal fees from Novartis, Kronos Bio, FibroGen and GV20 Therapeutics outside the submitted work. K. Cichowski reports other support from Erasca outside the submitted work. S.T. Barry reports employment and being a shareholder with AstraZeneca. R.R. Madsen reports grants from Wellcome Trust during the conduct of the study; as well as personal fees from Nested Therapeutics outside the submitted work. A. Toker reports grants from NCI and nonfinancial support from AstraZeneca during the conduct of the study; as well as personal fees from American Society for Biochemistry and Molecular Biology (ASBMB) and Novo Holdings of Novo Nordisk Foundation, and grants from BioHybrid Solutions outside the submitted work. No disclosures were reported by the other authors.
Figures
References
-
- Giaquinto AN, Sung H, Miller KD, Kramer JL, Newman LA, Minihan A, et al. . Breast cancer statistics, 2022. CA Cancer J Clin 2022;72:524–41. - PubMed
-
- Bianchini G, De Angelis C, Licata L, Gianni L. Treatment landscape of triple-negative breast cancer—expanded options, evolving needs. Nat Rev Clin Oncol 2022;19:91–113. - PubMed
MeSH terms
Substances
Grants and funding
- P01 CA250959/CA/NCI NIH HHS/United States
- no number/Orionin Tutkimussäätiö (Orion Research Foundation)
- DP-0194-21-00/Harvard Stem Cell Institute (HSCI)
- CCR18547665/Susan G. Komen (SGK)
- R01CA234600/National Institutes of Health (NIH)
- Challenge
- WT_/Wellcome Trust/United Kingdom
- no number/Ludwig Center at Harvard (Ludwig Center)
- 2020292129/National Science Foundation Graduate Research Fellowship Program (GRFP)
- R01 CA234600/CA/NCI NIH HHS/United States
- R35 CA253097/CA/NCI NIH HHS/United States
- CA253097/National Institutes of Health (NIH)
- W81XWH-20-1-0867/U.S. Department of Defense (DOD)
- no number/Howard Hughes Medical Institute (HHMI)
- P01CA250959/National Institutes of Health (NIH)
- no number/American Association for Cancer Research (AACR)
- no number/Maud Kuistilan Muistosäätiö (Maud Kuistila Foundation)
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
