

The splicing factor CCAR1 regulates the Fanconi anemia/BRCA pathway
- PMID: 39025073
- PMCID: PMC11321822
- DOI: 10.1016/j.molcel.2024.06.031
The splicing factor CCAR1 regulates the Fanconi anemia/BRCA pathway
Abstract
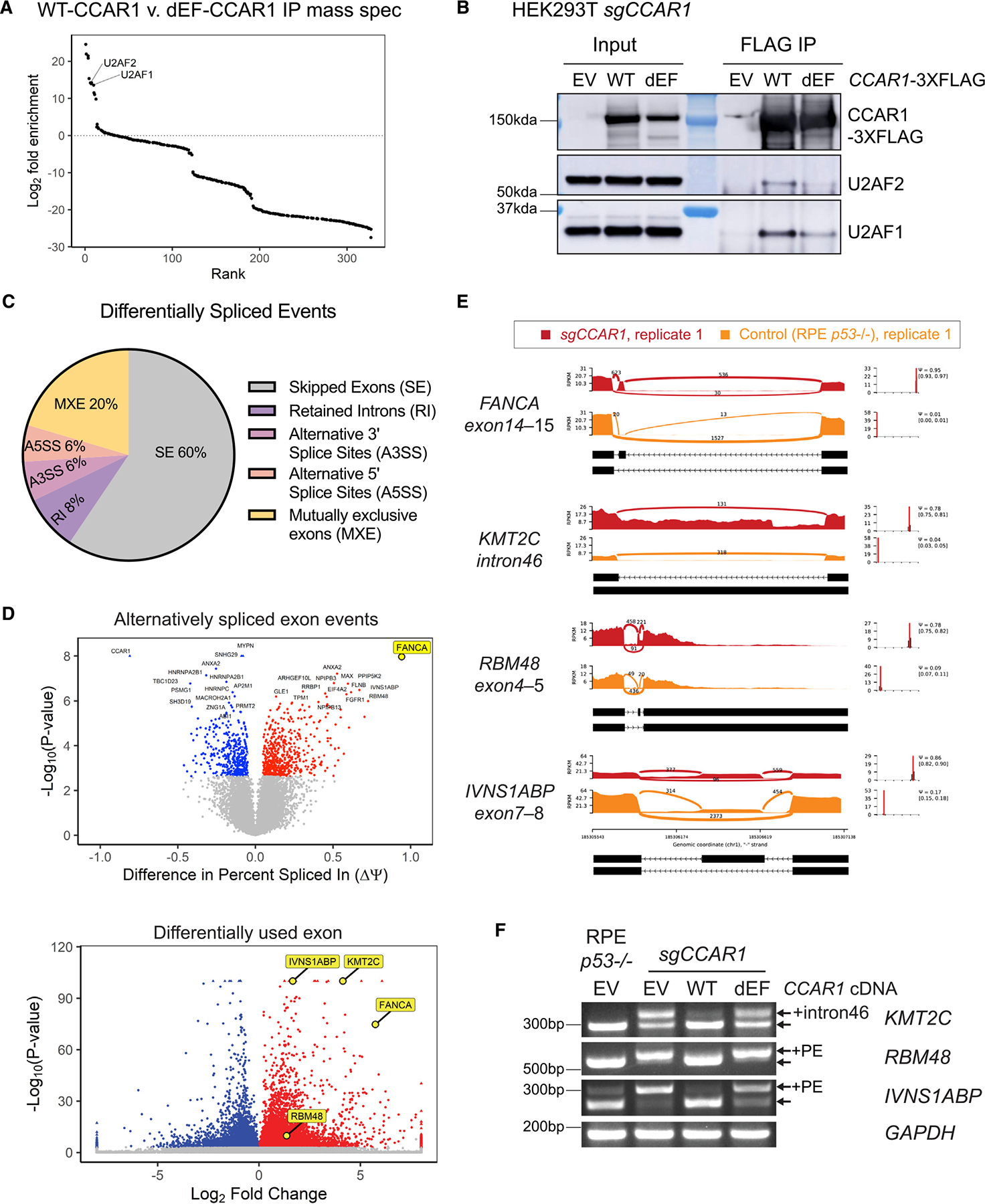
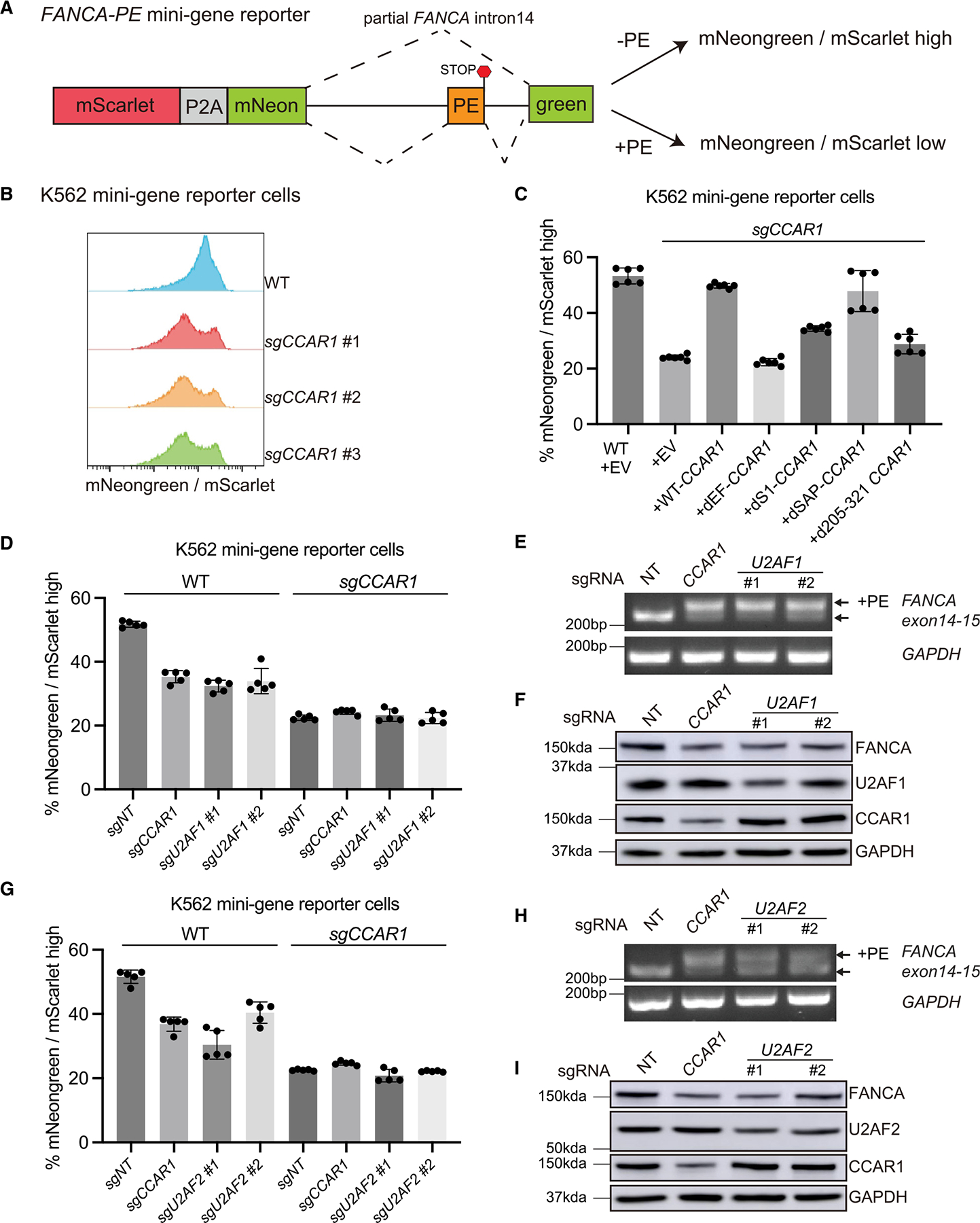
The twenty-three Fanconi anemia (FA) proteins cooperate in the FA/BRCA pathway to repair DNA interstrand cross-links (ICLs). The cell division cycle and apoptosis regulator 1 (CCAR1) protein is also a regulator of ICL repair, though its possible function in the FA/BRCA pathway remains unknown. Here, we demonstrate that CCAR1 plays a unique upstream role in the FA/BRCA pathway and is required for FANCA protein expression in human cells. Interestingly, CCAR1 co-immunoprecipitates with FANCA pre-mRNA and is required for FANCA mRNA processing. Loss of CCAR1 results in retention of a poison exon in the FANCA transcript, thereby leading to reduced FANCA protein expression. A unique domain of CCAR1, the EF hand domain, is required for interaction with the U2AF heterodimer of the spliceosome and for excision of the poison exon. Taken together, CCAR1 is a splicing modulator required for normal splicing of the FANCA mRNA and other mRNAs involved in various cellular pathways.
Keywords: CCAR1; DNA repair; EF hand; FANCA; Fanconi anemia; U2AF1/2; alternative splicing; poison exon.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests A.D.D. reports consulting for AbbVie, Deerfield Management Company, Impact Therapeutics, Moderna Therapeutics, PrimeFour Therapeutics, Schrödinger Inc., Servier BioInnovation LLC, and Tango Therapeutics; is a Scientific Advisory Board Member and Stockholder for Impact Therapeutics and Covant Therapeutics. K.A. is a member of the Advisory Board of Molecular Cell, the SAB of CAMP4 Therapeutics, consults for Syros Pharmaceuticals and Odyssey Therapeutics, and received research funding from Novartis not related to this work.
Figures
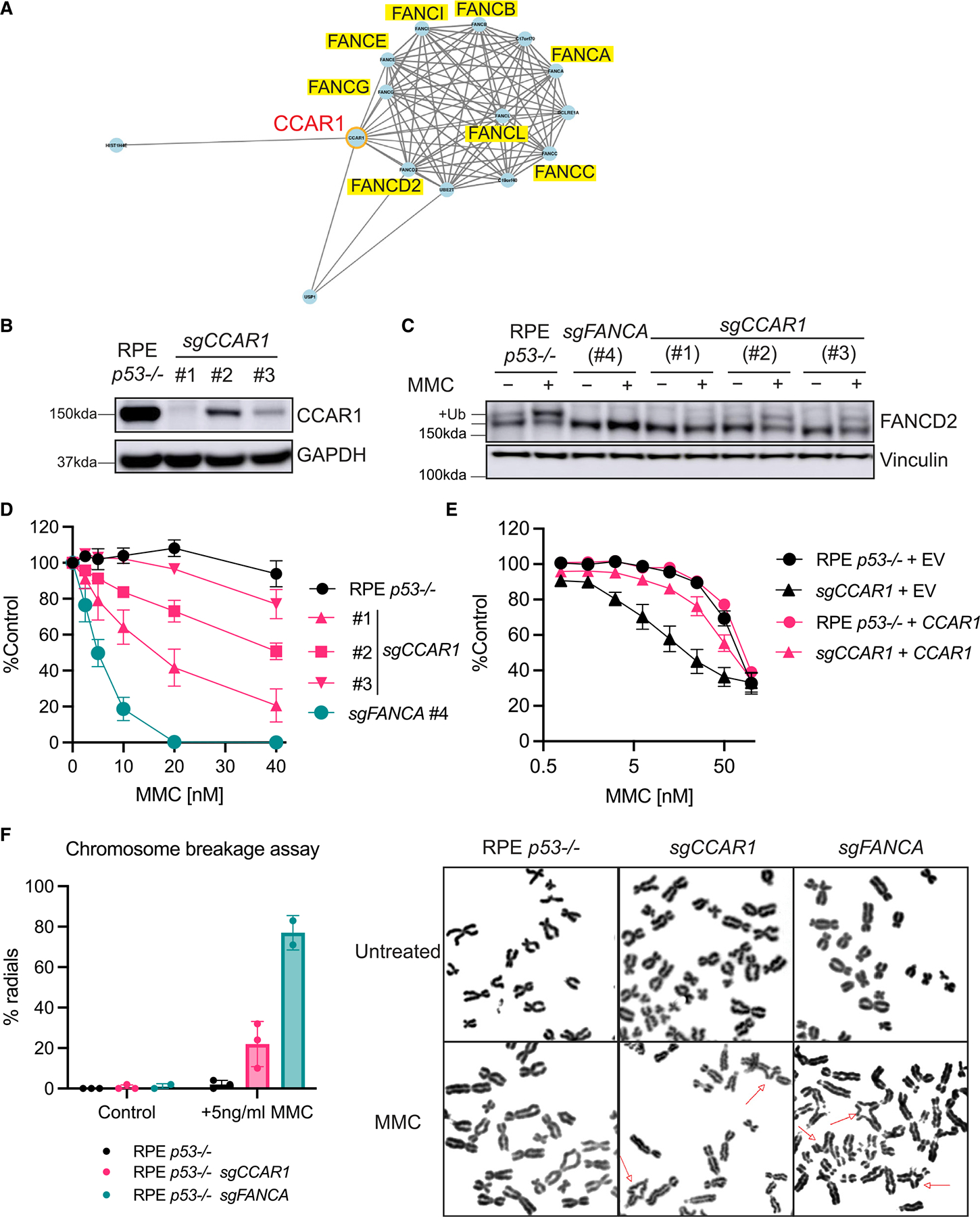
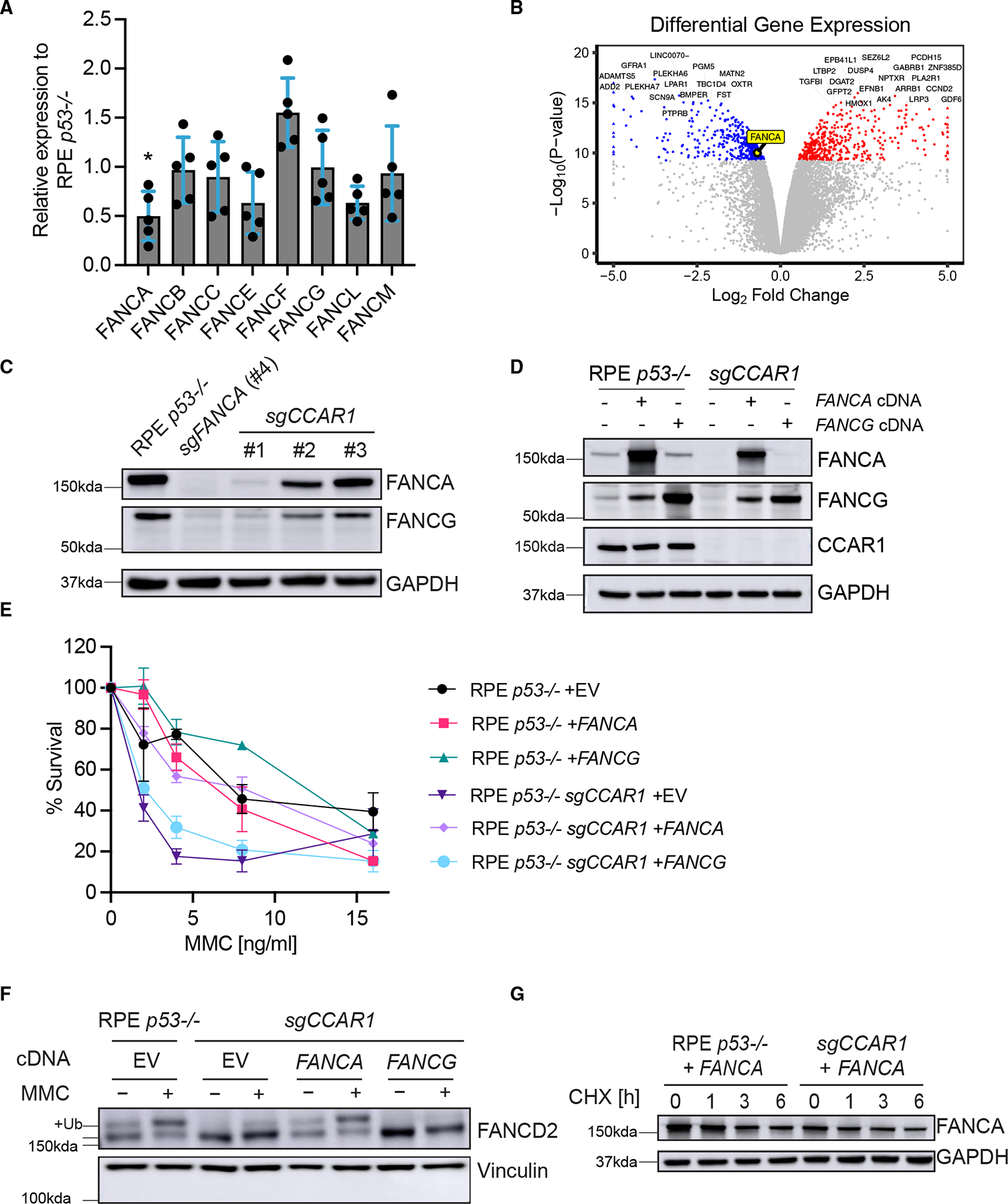
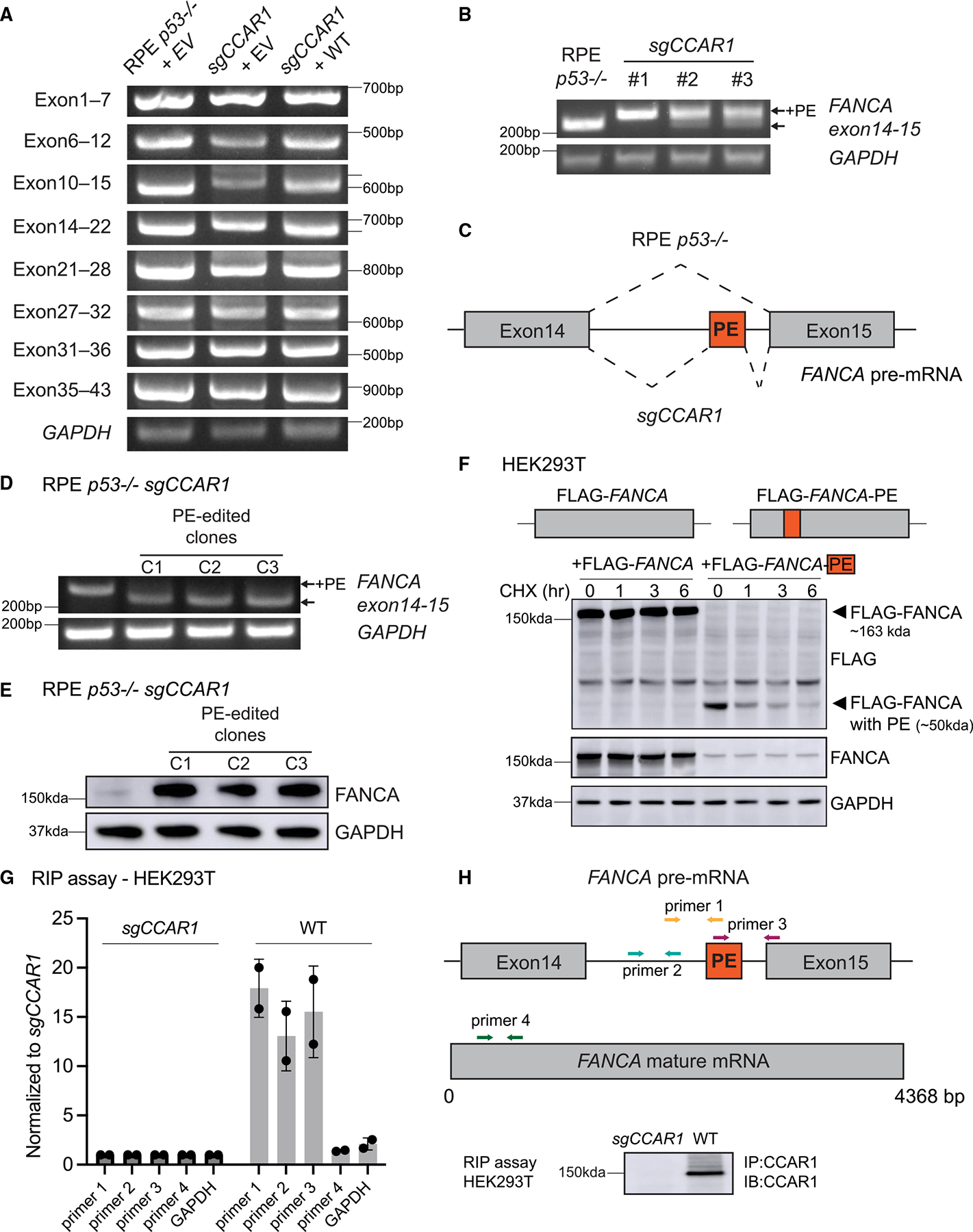
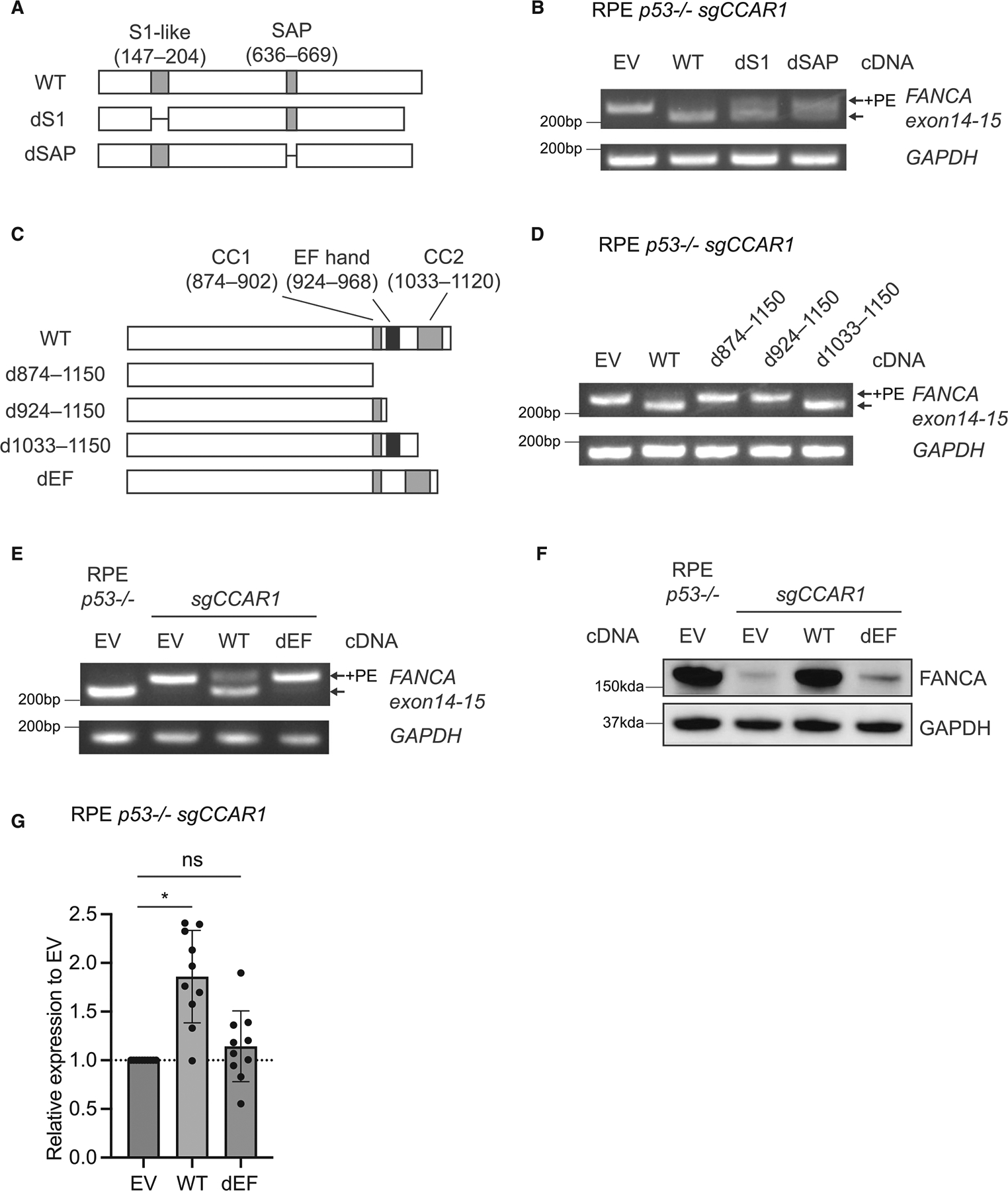
Comment in
-
Splicing regulation of DNA repair via CCAR1.Mol Cell. 2024 Jul 25;84(14):2591-2592. doi: 10.1016/j.molcel.2024.06.012. Mol Cell. 2024. PMID: 39059366
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
