

Symmetric and asymmetric DNA N6-adenine methylation regulates different biological responses in Mucorales
- PMID: 39025853
- PMCID: PMC11258239
- DOI: 10.1038/s41467-024-50365-2
Symmetric and asymmetric DNA N6-adenine methylation regulates different biological responses in Mucorales
Abstract
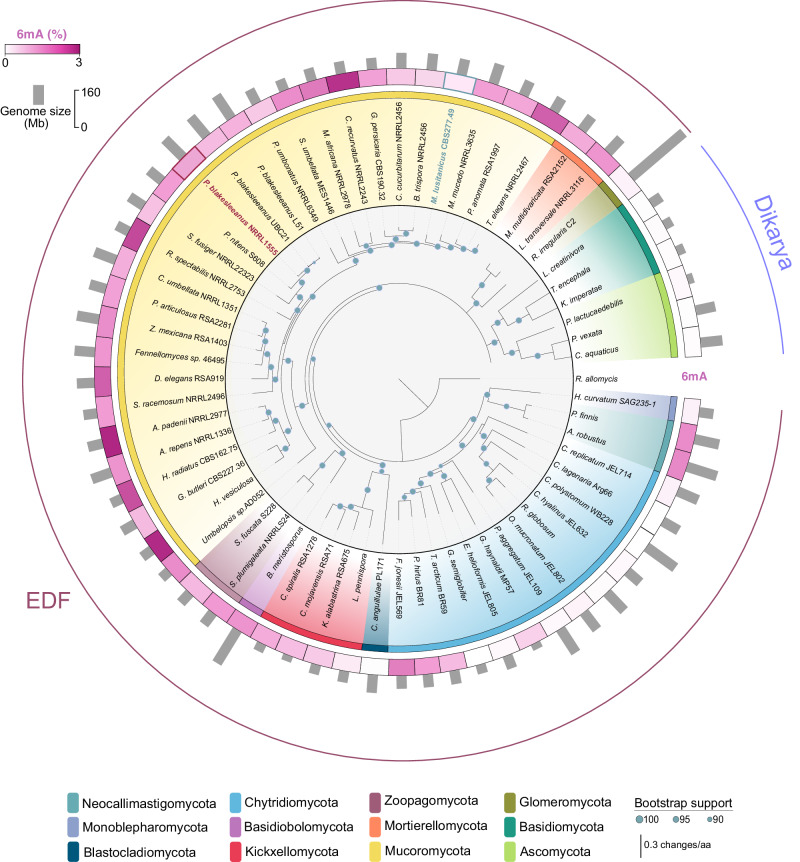
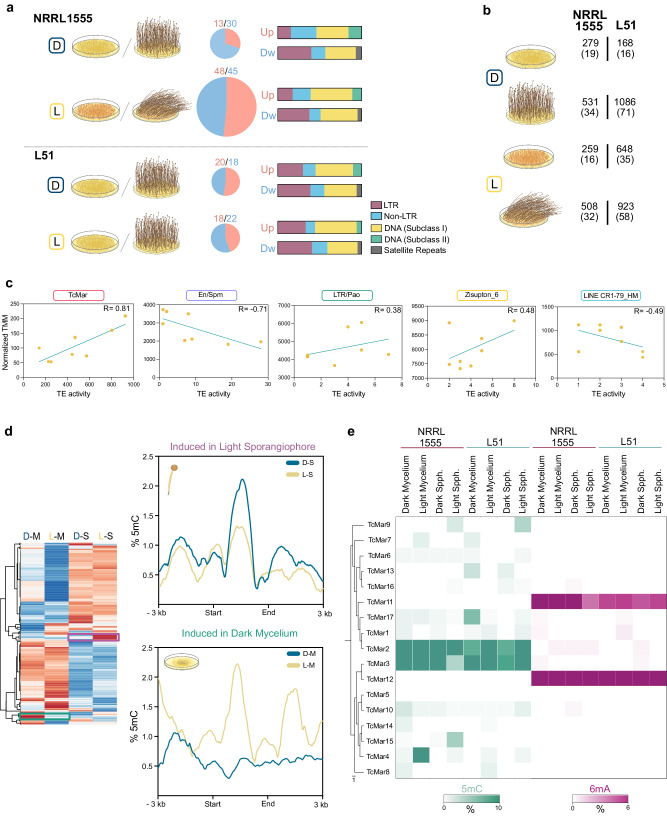
DNA N6-adenine methylation (6mA) has recently gained importance as an epigenetic modification in eukaryotes. Its function in lineages with high levels, such as early-diverging fungi (EDF), is of particular interest. Here, we investigated the biological significance and evolutionary implications of 6mA in EDF, which exhibit divergent evolutionary patterns in 6mA usage. The analysis of two Mucorales species displaying extreme 6mA usage reveals that species with high 6mA levels show symmetric methylation enriched in highly expressed genes. In contrast, species with low 6mA levels show mostly asymmetric 6mA. Interestingly, transcriptomic regulation throughout development and in response to environmental cues is associated with changes in the 6mA landscape. Furthermore, we identify an EDF-specific methyltransferase, likely originated from endosymbiotic bacteria, as responsible for asymmetric methylation, while an MTA-70 methylation complex performs symmetric methylation. The distinct phenotypes observed in the corresponding mutants reinforced the critical role of both types of 6mA in EDF.
© 2024. The Author(s).
Conflict of interest statement
The authors declare that they have not competing interests.
Figures





Similar articles
-
Symmetric adenine methylation is an essential DNA modification in the early-diverging fungus Rhizopus microsporus.Nat Commun. 2025 Apr 24;16(1):3843. doi: 10.1038/s41467-025-59170-x. Nat Commun. 2025. PMID: 40268918 Free PMC article.
-
Widespread adenine N6-methylation of active genes in fungi.Nat Genet. 2017 Jun;49(6):964-968. doi: 10.1038/ng.3859. Epub 2017 May 8. Nat Genet. 2017. PMID: 28481340
-
A distinct class of eukaryotic MT-A70 methyltransferases maintain symmetric DNA N6-adenine methylation at the ApT dinucleotides as an epigenetic mark associated with transcription.Nucleic Acids Res. 2019 Dec 16;47(22):11771-11789. doi: 10.1093/nar/gkz1053. Nucleic Acids Res. 2019. PMID: 31722409 Free PMC article.
-
Epigenetic Methylations on N6-Adenine and N6-Adenosine with the same Input but Different Output.Int J Mol Sci. 2019 Jun 15;20(12):2931. doi: 10.3390/ijms20122931. Int J Mol Sci. 2019. PMID: 31208067 Free PMC article. Review.
-
The exploration of N6-deoxyadenosine methylation in mammalian genomes.Protein Cell. 2021 Oct;12(10):756-768. doi: 10.1007/s13238-021-00866-3. Epub 2021 Aug 17. Protein Cell. 2021. PMID: 34405377 Free PMC article. Review.
Cited by
-
Future Prospects and Challenges in Mucormycosis Research.J Fungi (Basel). 2025 Jul 22;11(8):545. doi: 10.3390/jof11080545. J Fungi (Basel). 2025. PMID: 40863497 Free PMC article.
-
SMAC: identifying DNA N6-methyladenine (6mA) at the single-molecule level using SMRT CCS data.Brief Bioinform. 2025 Mar 4;26(2):bbaf153. doi: 10.1093/bib/bbaf153. Brief Bioinform. 2025. PMID: 40205850 Free PMC article.
-
Consecutive low-frequency shifts in A/T content denote nucleosome positions across microeukaryotes.iScience. 2025 Apr 18;28(5):112472. doi: 10.1016/j.isci.2025.112472. eCollection 2025 May 16. iScience. 2025. PMID: 40491964 Free PMC article.
-
Symmetric adenine methylation is an essential DNA modification in the early-diverging fungus Rhizopus microsporus.Nat Commun. 2025 Apr 24;16(1):3843. doi: 10.1038/s41467-025-59170-x. Nat Commun. 2025. PMID: 40268918 Free PMC article.
-
Repressive Cytosine Methylation is a Marker of Viral Gene Transfer Across Divergent Eukaryotes.Mol Biol Evol. 2025 Jul 30;42(8):msaf176. doi: 10.1093/molbev/msaf176. Mol Biol Evol. 2025. PMID: 40712095 Free PMC article.
References
-
- Wijayawardene N. Outline of Fungi and fungus-like taxa. Mycosphere. 2020;11:1060–1456. doi: 10.5943/mycosphere/11/1/8. - DOI
-
- Berbee, M. L. & Taylor, J. W. The mycota VIIA. In Systematics and Evolution. 229–245 (Springer, Berlin, Heidelberg, 2001)
MeSH terms
Substances
Grants and funding
- PID2021-124674NB-I00/Ministry of Economy and Competitiveness | Agencia Estatal de Investigación (Spanish Agencia Estatal de Investigación)
- PID2021-128001OB-I00/Ministry of Economy and Competitiveness | Agencia Estatal de Investigación (Spanish Agencia Estatal de Investigación)
- 20897/PI/18/Fundación Séneca (Fundacion Seneca)
- 21969/PI/22/Fundación Séneca (Fundacion Seneca)
- P20-00622/Junta de Andalucía
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
