

Integrating muti-omics data to identify tissue-specific DNA methylation biomarkers for cancer risk
- PMID: 39025880
- PMCID: PMC11258330
- DOI: 10.1038/s41467-024-50404-y
Integrating muti-omics data to identify tissue-specific DNA methylation biomarkers for cancer risk
Abstract
The relationship between tissue-specific DNA methylation and cancer risk remains inadequately elucidated. Leveraging resources from the Genotype-Tissue Expression consortium, here we develop genetic models to predict DNA methylation at CpG sites across the genome for seven tissues and apply these models to genome-wide association study data of corresponding cancers, namely breast, colorectal, renal cell, lung, ovarian, prostate, and testicular germ cell cancers. At Bonferroni-corrected P < 0.05, we identify 4248 CpGs that are significantly associated with cancer risk, of which 95.4% (4052) are specific to a particular cancer type. Notably, 92 CpGs within 55 putative novel loci retain significant associations with cancer risk after conditioning on proximal signals identified by genome-wide association studies. Integrative multi-omics analyses reveal 854 CpG-gene-cancer trios, suggesting that DNA methylation at 309 distinct CpGs might influence cancer risk through regulating the expression of 205 unique cis-genes. These findings substantially advance our understanding of the interplay between genetics, epigenetics, and gene expression in cancer etiology.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
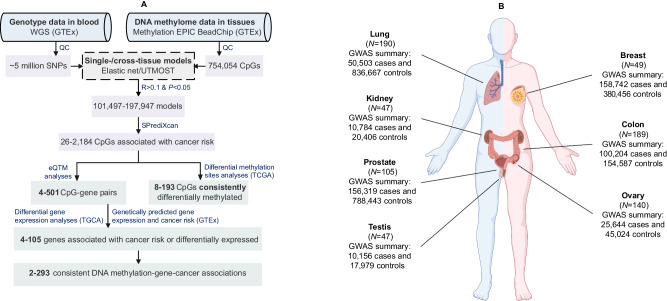
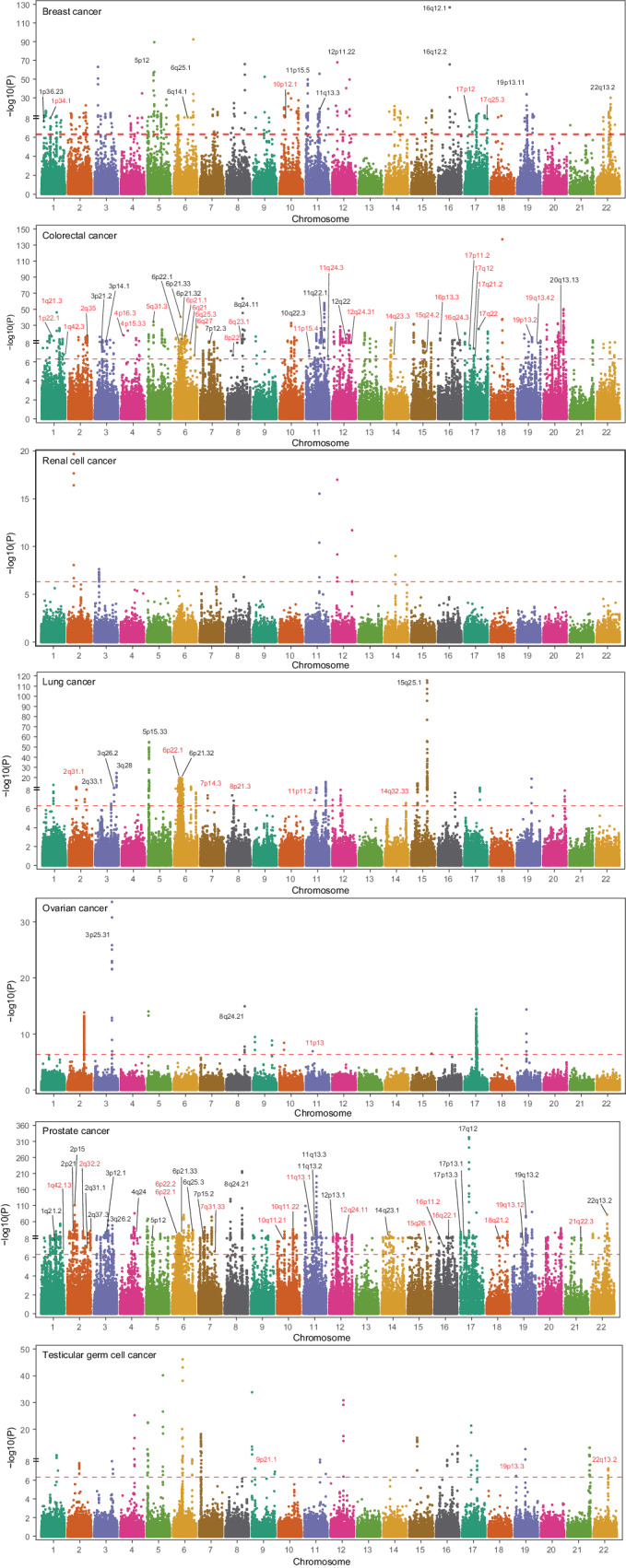
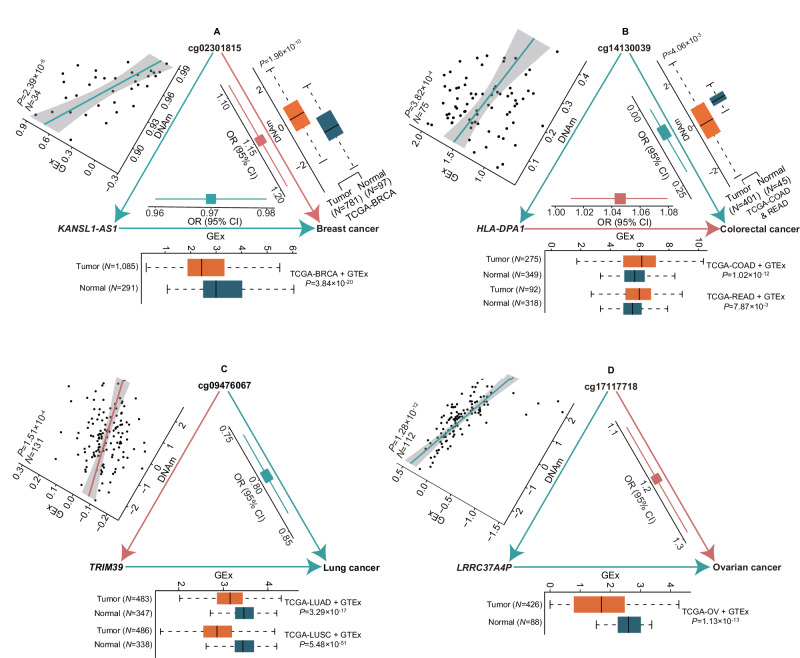
References
MeSH terms
Substances
Supplementary concepts
Grants and funding
- R01 CA247987/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- R01CA247987/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R01CA249863/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R00CA248822/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
LinkOut - more resources
Full Text Sources
Medical
