

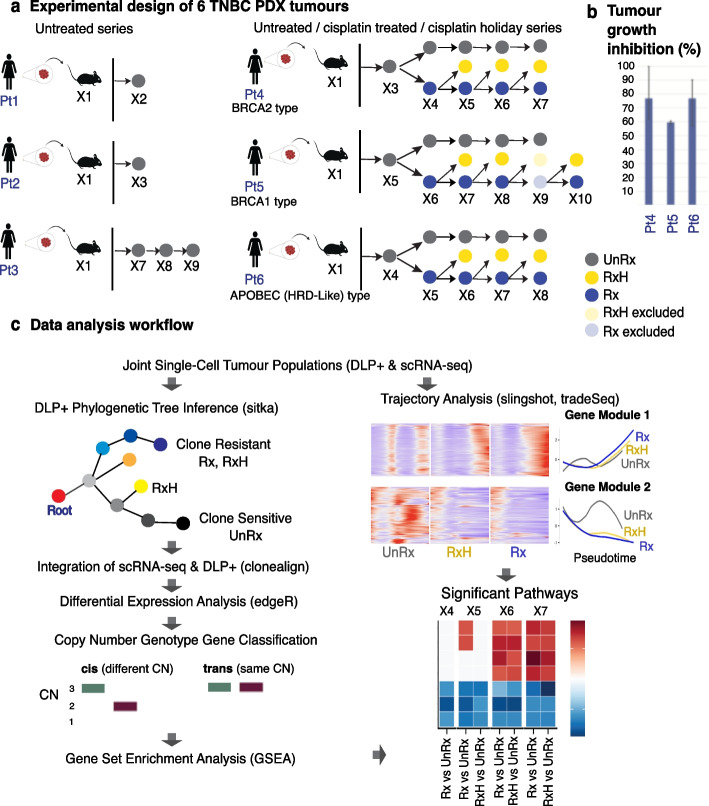
Single-cell decoding of drug induced transcriptomic reprogramming in triple negative breast cancers
- PMID: 39026273
- PMCID: PMC11256464
- DOI: 10.1186/s13059-024-03318-3
Single-cell decoding of drug induced transcriptomic reprogramming in triple negative breast cancers
Abstract
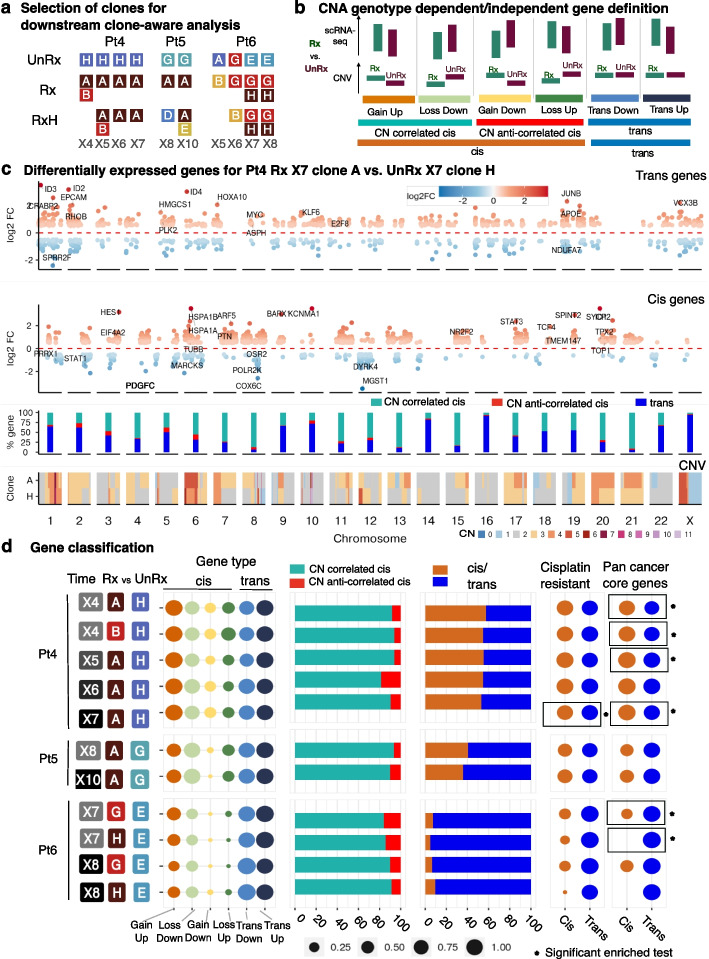
Background: The encoding of cell intrinsic drug resistance states in breast cancer reflects the contributions of genomic and non-genomic variations and requires accurate estimation of clonal fitness from co-measurement of transcriptomic and genomic data. Somatic copy number (CN) variation is the dominant mutational mechanism leading to transcriptional variation and notably contributes to platinum chemotherapy resistance cell states. Here, we deploy time series measurements of triple negative breast cancer (TNBC) single-cell transcriptomes, along with co-measured single-cell CN fitness, identifying genomic and transcriptomic mechanisms in drug-associated transcriptional cell states.
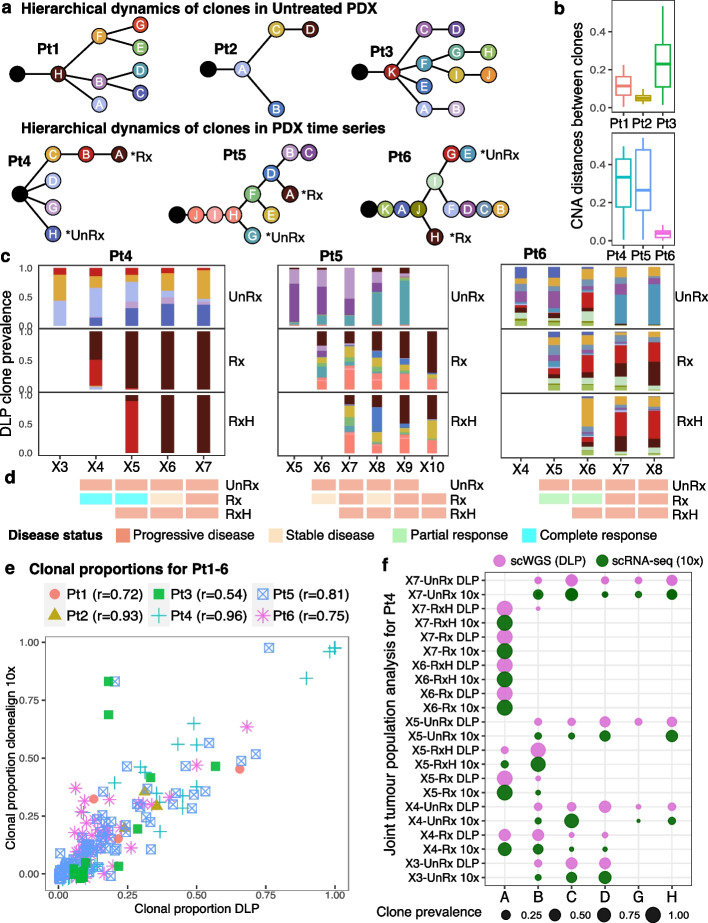
Results: We present scRNA-seq data (53,641 filtered cells) from serial passaging TNBC patient-derived xenograft (PDX) experiments spanning 2.5 years, matched with genomic single-cell CN data from the same samples. Our findings reveal distinct clonal responses within TNBC tumors exposed to platinum. Clones with high drug fitness undergo clonal sweeps and show subtle transcriptional reversion, while those with weak fitness exhibit dynamic transcription upon drug withdrawal. Pathway analysis highlights convergence on epithelial-mesenchymal transition and cytokine signaling, associated with resistance. Furthermore, pseudotime analysis demonstrates hysteresis in transcriptional reversion, indicating generation of new intermediate transcriptional states upon platinum exposure.
Conclusions: Within a polyclonal tumor, clones with strong genotype-associated fitness under platinum remained fixed, minimizing transcriptional reversion upon drug withdrawal. Conversely, clones with weaker fitness display non-genomic transcriptional plasticity. This suggests CN-associated and CN-independent transcriptional states could both contribute to platinum resistance. The dominance of genomic or non-genomic mechanisms within polyclonal tumors has implications for drug sensitivity, restoration, and re-treatment strategies.
Keywords: Cisplatin treatment; Clone aware analysis; DLP+ single-cell sequencing; In-cis/in-trans genes; PDX; Sensitive/resistant clones; Single-cell RNA sequencing.
© 2024. The Author(s).
Conflict of interest statement
Samuel Aparicio is founder of Genome Therapeutics Ltd, outside the scope of this study. The remaining authors declare that they have no competing interests.
Figures



References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
