

Hem1 inborn errors of immunity: waving goodbye to coordinated immunity in mice and humans
- PMID: 39026677
- PMCID: PMC11254771
- DOI: 10.3389/fimmu.2024.1402139
Hem1 inborn errors of immunity: waving goodbye to coordinated immunity in mice and humans
Abstract
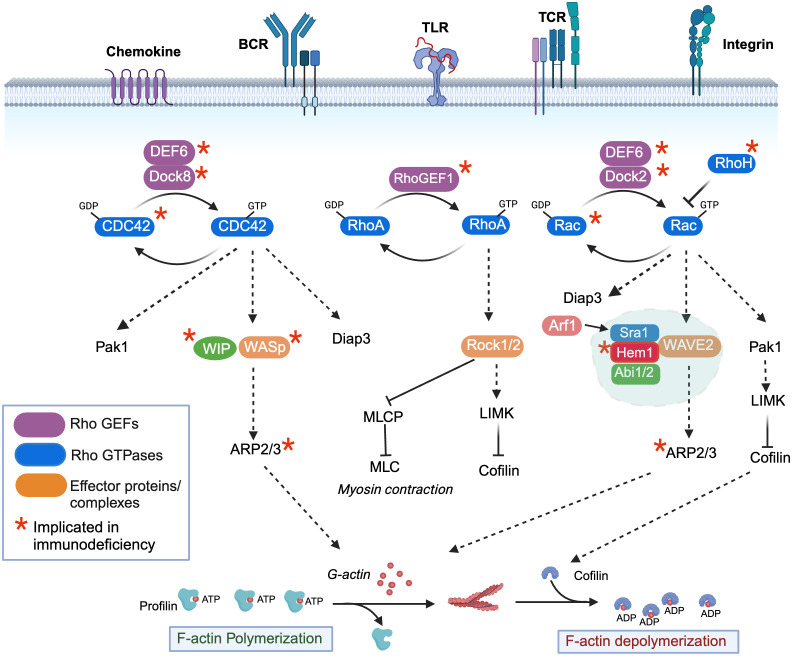
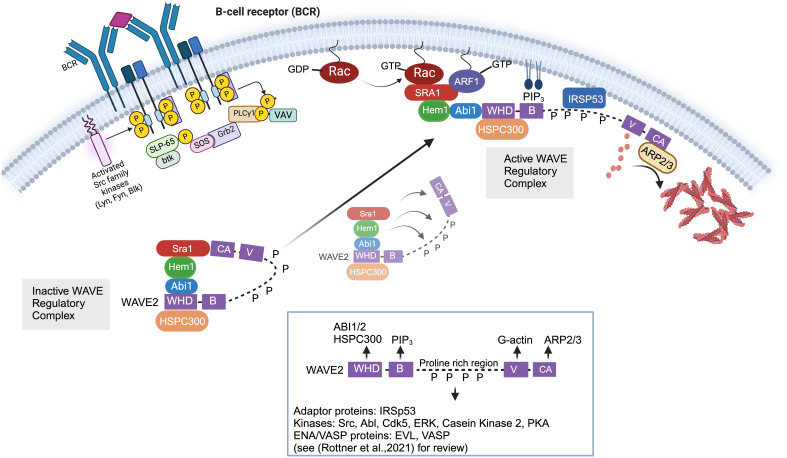
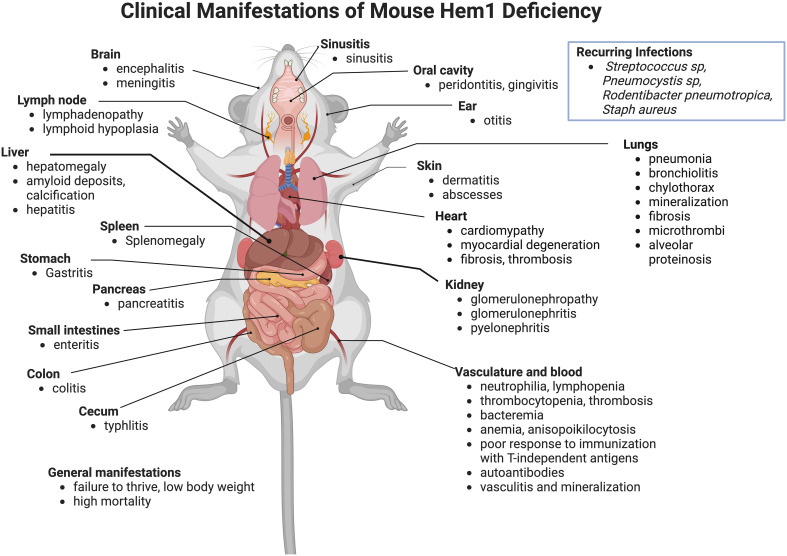
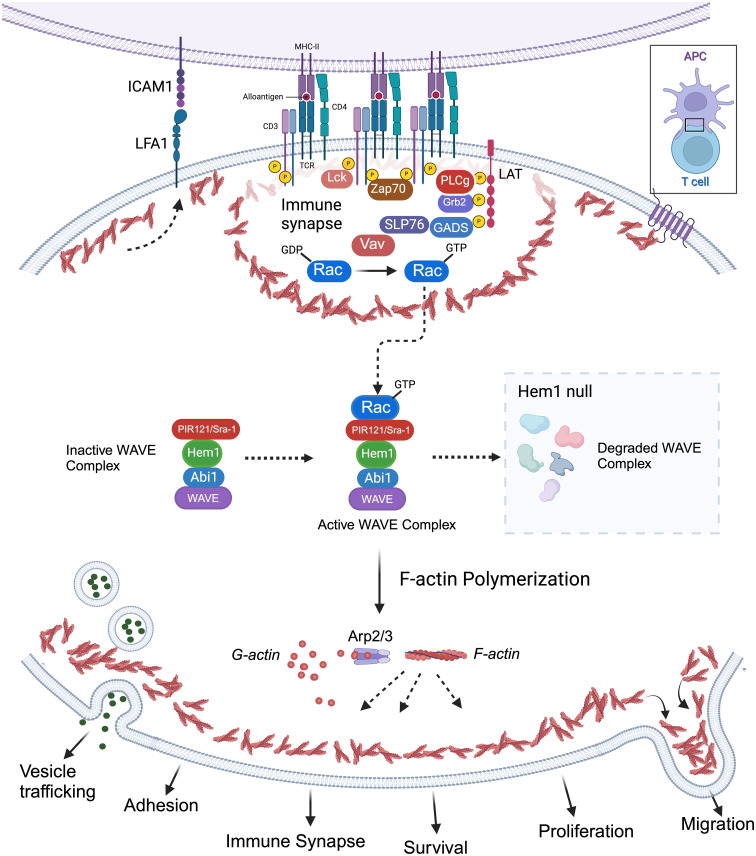
Inborn errors of immunity (IEI) are a group of diseases in humans that typically present as increased susceptibility to infections, autoimmunity, hyperinflammation, allergy, and in some cases malignancy. Among newly identified genes linked to IEIs include 3 independent reports of 9 individuals from 7 independent kindreds with severe primary immunodeficiency disease (PID) and autoimmunity due to loss-of-function mutations in the NCKAP1L gene encoding Hematopoietic protein 1 (HEM1). HEM1 is a hematopoietic cell specific component of the WASp family verprolin homologous (WAVE) regulatory complex (WRC), which acts downstream of multiple immune receptors to stimulate actin nucleation and polymerization of filamentous actin (F-actin). The polymerization and branching of F-actin is critical for creating force-generating cytoskeletal structures which drive most active cellular processes including migration, adhesion, immune synapse formation, and phagocytosis. Branched actin networks at the cell cortex have also been implicated in acting as a barrier to regulate inappropriate vesicle (e.g. cytokine) secretion and spontaneous antigen receptor crosslinking. Given the importance of the actin cytoskeleton in most or all hematopoietic cells, it is not surprising that HEM1 deficient children present with a complex clinical picture that involves overlapping features of immunodeficiency and autoimmunity. In this review, we will provide an overview of what is known about the molecular and cellular functions of HEM1 and the WRC in immune and other cells. We will describe the common clinicopathological features and immunophenotypes of HEM1 deficiency in humans and provide detailed comparative descriptions of what has been learned about Hem1 disruption using constitutive and immune cell-specific mouse knockout models. Finally, we discuss future perspectives and important areas for investigation regarding HEM1 and the WRC.
Keywords: Hem-1; Hem1; NCKAP1L; actin cytoskeleton; hematopoietic protein-1; immunodeficiency disease; inborn errors of immunity; wave.
Copyright © 2024 Christodoulou, Tsai, Suwankitwat, Anderson and Iritani.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
HEM1 Actin Immunodysregulatory Disorder: Genotypes, Phenotypes, and Future Directions.J Clin Immunol. 2022 Nov;42(8):1583-1592. doi: 10.1007/s10875-022-01327-0. Epub 2022 Jul 22. J Clin Immunol. 2022. PMID: 35869404 Free PMC article. Review.
-
HEM1 deficiency disrupts mTORC2 and F-actin control in inherited immunodysregulatory disease.Science. 2020 Jul 10;369(6500):202-207. doi: 10.1126/science.aay5663. Science. 2020. PMID: 32647003 Free PMC article.
-
Hem1 controls T cell activation, memory, and the regulated release of immunosuppressive and proinflammatory cytokines.JCI Insight. 2025 Jul 8:e174235. doi: 10.1172/jci.insight.174235. Online ahead of print. JCI Insight. 2025. PMID: 40627451
-
The cytoskeletal regulator HEM1 governs B cell development and prevents autoimmunity.Sci Immunol. 2020 Jul 10;5(49):eabc3979. doi: 10.1126/sciimmunol.abc3979. Sci Immunol. 2020. PMID: 32646852 Free PMC article.
-
Hem-1: putting the "WAVE" into actin polymerization during an immune response.FEBS Lett. 2010 Dec 15;584(24):4923-32. doi: 10.1016/j.febslet.2010.10.018. Epub 2010 Oct 20. FEBS Lett. 2010. PMID: 20969869 Free PMC article. Review.
Cited by
-
The WAVE complex in developmental and adulthood brain disorders.Exp Mol Med. 2025 Feb;57(1):13-29. doi: 10.1038/s12276-024-01386-w. Epub 2025 Jan 7. Exp Mol Med. 2025. PMID: 39774290 Free PMC article. Review.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials