

ASF1A-dependent P300-mediated histone H3 lysine 18 lactylation promotes atherosclerosis by regulating EndMT
- PMID: 39027248
- PMCID: PMC11252488
- DOI: 10.1016/j.apsb.2024.03.008
ASF1A-dependent P300-mediated histone H3 lysine 18 lactylation promotes atherosclerosis by regulating EndMT
Abstract
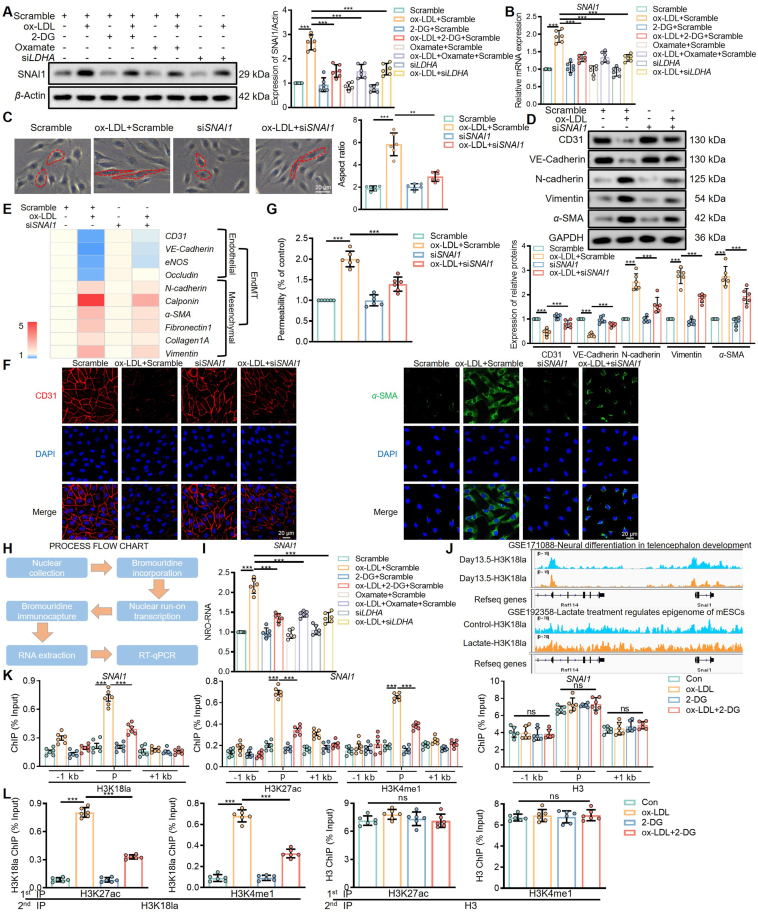
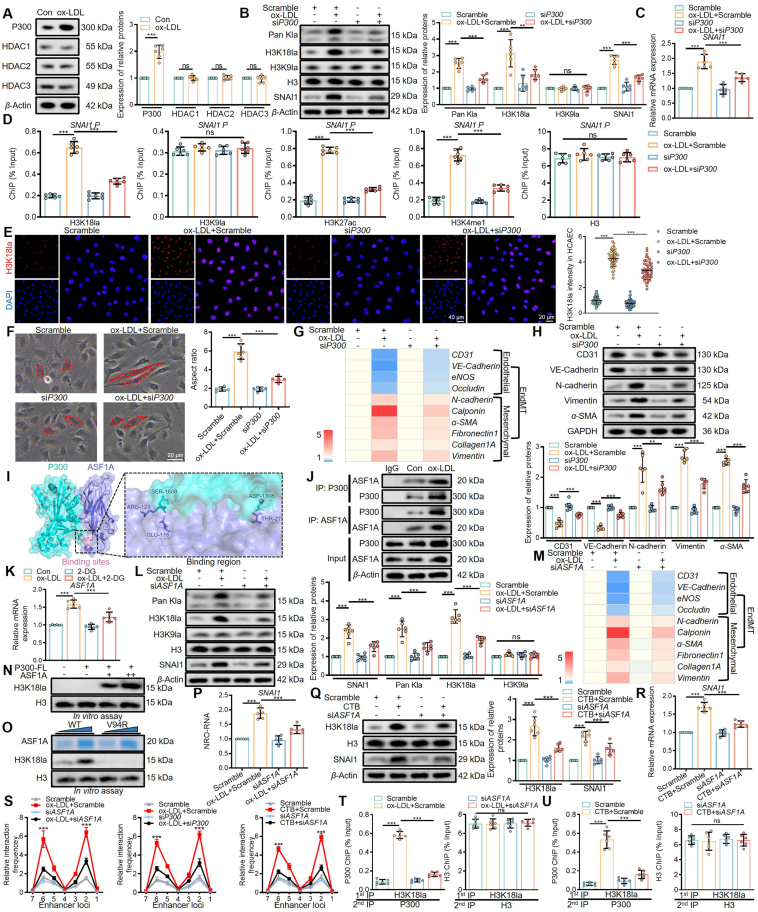
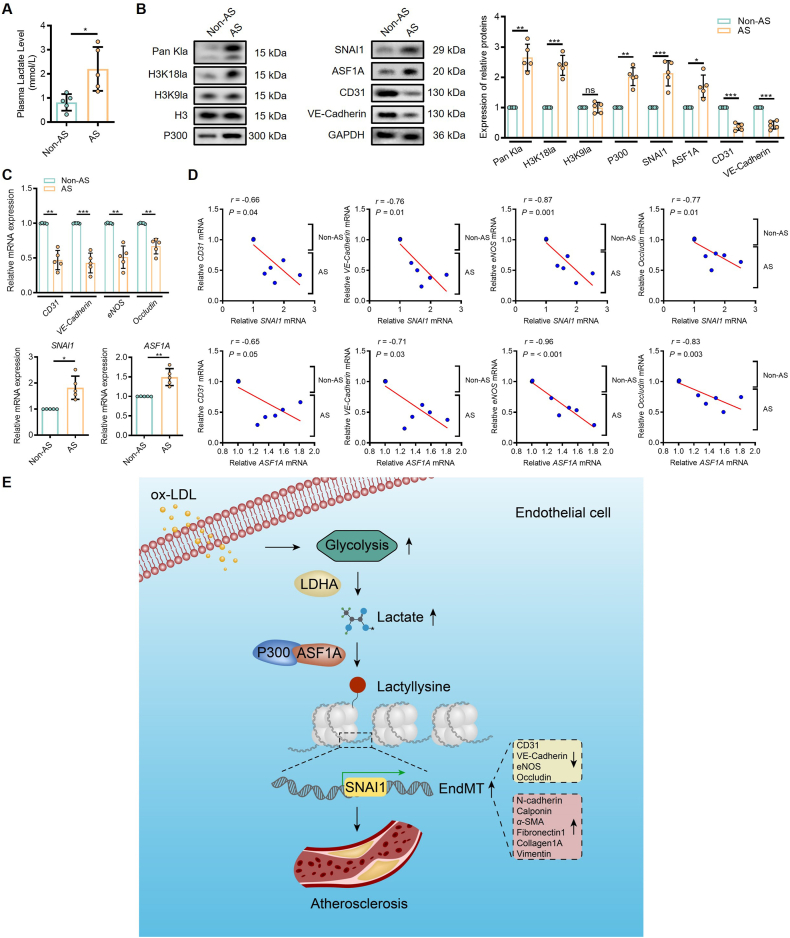
Endothelial-to-mesenchymal transition (EndMT) is a key driver of atherosclerosis. Aerobic glycolysis is increased in the endothelium of atheroprone areas, accompanied by elevated lactate levels. Histone lactylation, mediated by lactate, can regulate gene expression and participate in disease regulation. However, whether histone lactylation is involved in atherosclerosis remains unknown. Here, we report that lipid peroxidation could lead to EndMT-induced atherosclerosis by increasing lactate-dependent histone H3 lysine 18 lactylation (H3K18la) in vitro and in vivo, as well as in atherosclerotic patients' arteries. Mechanistically, the histone chaperone ASF1A was first identified as a cofactor of P300, which precisely regulated the enrichment of H3K18la at the promoter of SNAI1, thereby activating SNAI1 transcription and promoting EndMT. We found that deletion of ASF1A inhibited EndMT and improved endothelial dysfunction. Functional analysis based on Apoe KO Asf1a ECKO mice in the atherosclerosis model confirmed the involvement of H3K18la in atherosclerosis and found that endothelium-specific ASF1A deficiency inhibited EndMT and alleviated atherosclerosis development. Inhibition of glycolysis by pharmacologic inhibition and advanced PROTAC attenuated H3K18la, SNAI1 transcription, and EndMT-induced atherosclerosis. This study illustrates precise crosstalk between metabolism and epigenetics via H3K18la by the P300/ASF1A molecular complex during EndMT-induced atherogenesis, which provides emerging therapies for atherosclerosis.
Keywords: ASF1A; Atherosclerosis; Endothelial dysfunction; Endothelial-to-mesenchymal transition; Epigenetic; Histone lactylation; Lactate; SNAI1.
© 2024 The Authors.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures






References
-
- Herrington W., Lacey B., Sherliker P., Armitage J., Lewington S. Epidemiology of atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ Res. 2016;118:535–546. - PubMed
-
- Xu S., Ilyas I., Little P.J., Li H., Kamato D., Zheng X., et al. Endothelial dysfunction in atherosclerotic cardiovascular diseases and beyond: from mechanism to pharmacotherapies. Pharmacol Rev. 2021;73:924–967. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
