

Unraveling neurovascular mysteries: the role of endothelial glycocalyx dysfunction in Alzheimer's disease pathogenesis
- PMID: 39027900
- PMCID: PMC11254711
- DOI: 10.3389/fphys.2024.1394725
Unraveling neurovascular mysteries: the role of endothelial glycocalyx dysfunction in Alzheimer's disease pathogenesis
Abstract
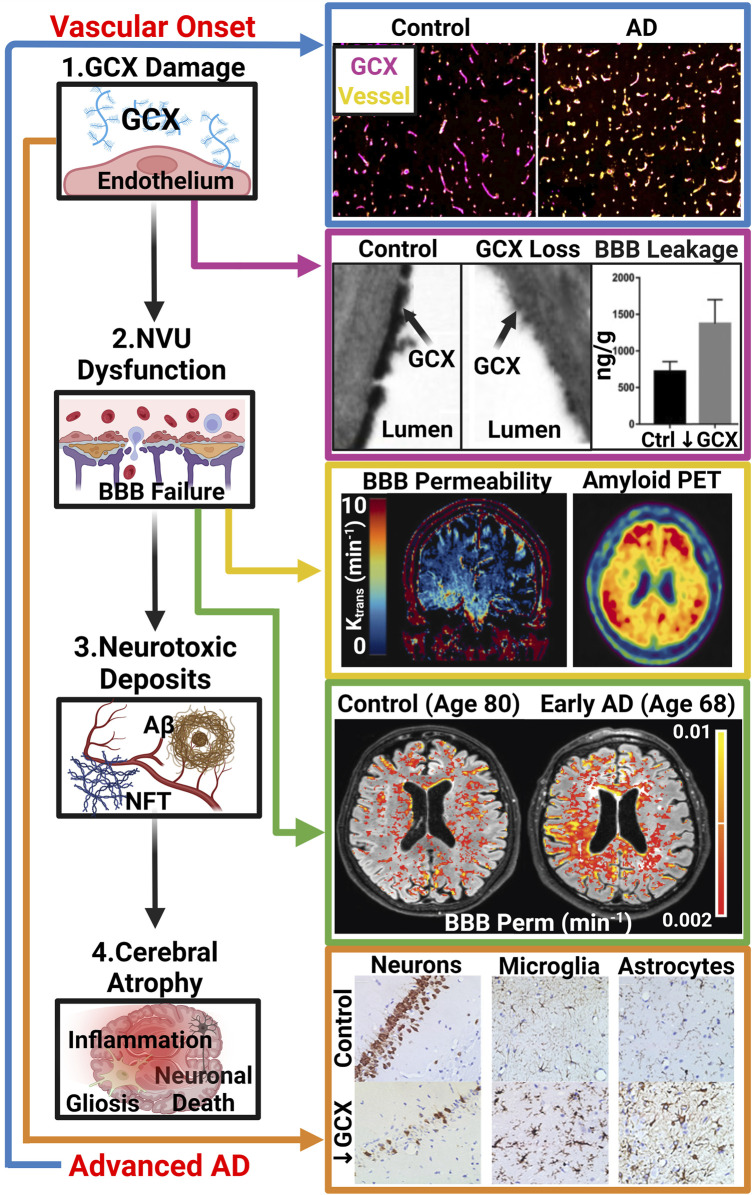
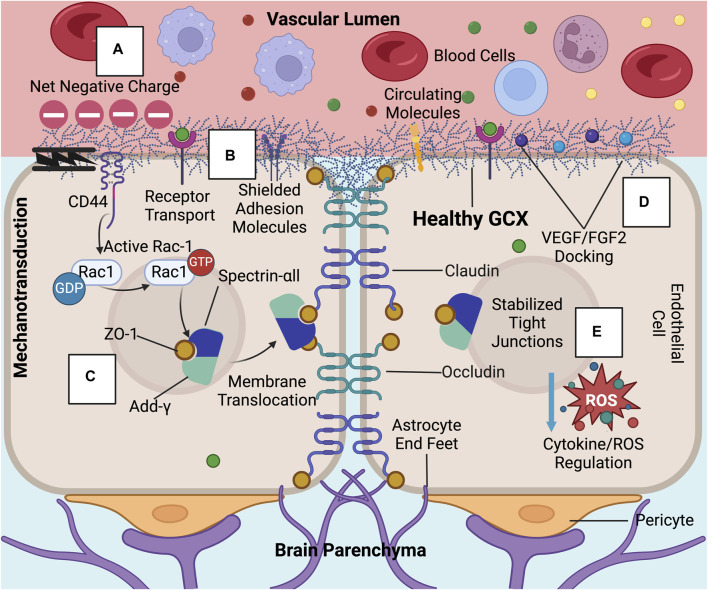
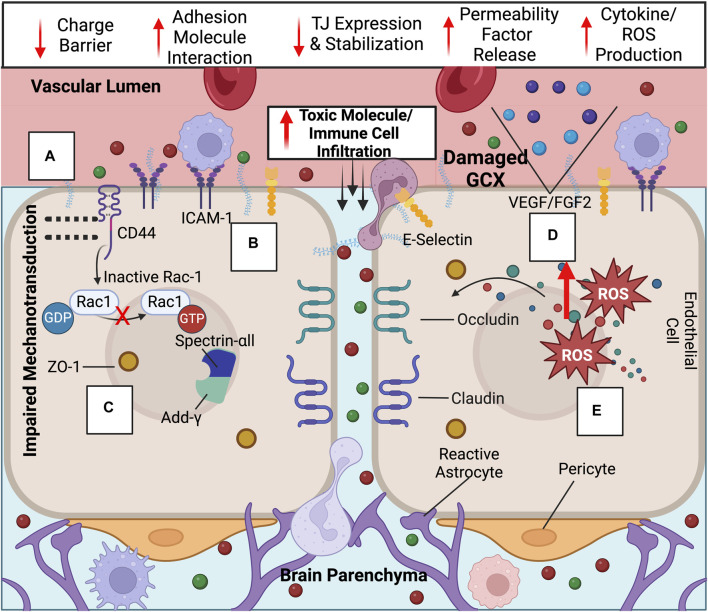
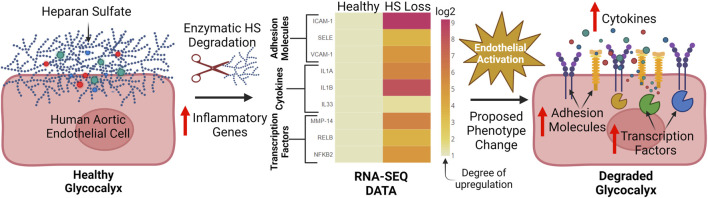
While cardiovascular disease, cancer, and human immunodeficiency virus (HIV) mortality rates have decreased over the past 20 years, Alzheimer's Disease (AD) deaths have risen by 145% since 2010. Despite significant research efforts, effective AD treatments remain elusive due to a poorly defined etiology and difficulty in targeting events that occur too downstream of disease onset. In hopes of elucidating alternative treatment pathways, now, AD is commonly being more broadly defined not only as a neurological disorder but also as a progression of a variety of cerebrovascular pathologies highlighted by the breakdown of the blood-brain barrier. The endothelial glycocalyx (GCX), which is an essential regulator of vascular physiology, plays a crucial role in the function of the neurovascular system, acting as an essential vascular mechanotransducer to facilitate ultimate blood-brain homeostasis. Shedding of the cerebrovascular GCX could be an early indication of neurovascular dysfunction and may subsequently progress neurodegenerative diseases like AD. Recent advances in in vitro modeling, gene/protein silencing, and imaging techniques offer new avenues of scrutinizing the GCX's effects on AD-related neurovascular pathology. Initial studies indicate GCX degradation in AD and other neurodegenerative diseases and have begun to demonstrate a possible link to GCX loss and cerebrovascular dysfunction. This review will scrutinize the GCX's contribution to known vascular etiologies of AD and propose future work aimed at continuing to uncover the relationship between GCX dysfunction and eventual AD-associated neurological deterioration.
Keywords: Alzheimer’s disease; blood-brain barrier; endothelial glycocalyx; neurovascular dysfunction; vascular etiology; vascular mechanobiology.
Copyright © 2024 O’Hare, Millican and Ebong.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Evidence of endothelial dysfunction in the development of Alzheimer's disease: Is Alzheimer's a vascular disorder?Am J Cardiovasc Dis. 2013 Nov 1;3(4):197-226. Am J Cardiovasc Dis. 2013. PMID: 24224133 Free PMC article. Review.
-
Diosmin and its glycocalyx restorative and anti-inflammatory effects on injured blood vessels.FASEB J. 2022 Dec;36(12):e22630. doi: 10.1096/fj.202200053RR. FASEB J. 2022. PMID: 36315163
-
Glycocalyx is critical for blood-brain barrier integrity by suppressing caveolin1-dependent endothelial transcytosis following ischemic stroke.Brain Pathol. 2022 Jan;32(1):e13006. doi: 10.1111/bpa.13006. Epub 2021 Jul 19. Brain Pathol. 2022. PMID: 34286899 Free PMC article.
-
Endothelial barrier reinforcement relies on flow-regulated glycocalyx, a potential therapeutic target.Biorheology. 2019;56(2-3):131-149. doi: 10.3233/BIR-180205. Biorheology. 2019. PMID: 30988234 Free PMC article. Review.
-
Glycocalyx in Atherosclerosis-Relevant Endothelium Function and as a Therapeutic Target.Curr Atheroscler Rep. 2017 Nov 10;19(12):63. doi: 10.1007/s11883-017-0691-9. Curr Atheroscler Rep. 2017. PMID: 29127504 Free PMC article. Review.
Cited by
-
Sweet Aging: Glycocalyx and Galectins in CNS Aging and Neurodegenerative Disorders.Int J Mol Sci. 2025 May 14;26(10):4699. doi: 10.3390/ijms26104699. Int J Mol Sci. 2025. PMID: 40429840 Free PMC article. Review.
-
Traumatic Brain Injury and Alzheimer's Disease: A Shared Neurovascular Hypothesis.Neurosci Insights. 2025 Mar 20;20:26331055251323292. doi: 10.1177/26331055251323292. eCollection 2025. Neurosci Insights. 2025. PMID: 40124421 Free PMC article. Review.
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous