

Cellular and molecular mechanisms of the blood-brain barrier dysfunction in neurodegenerative diseases
- PMID: 39030617
- PMCID: PMC11264766
- DOI: 10.1186/s12987-024-00557-1
Cellular and molecular mechanisms of the blood-brain barrier dysfunction in neurodegenerative diseases
Abstract
Background: Maintaining the structural and functional integrity of the blood-brain barrier (BBB) is vital for neuronal equilibrium and optimal brain function. Disruptions to BBB performance are implicated in the pathology of neurodegenerative diseases.
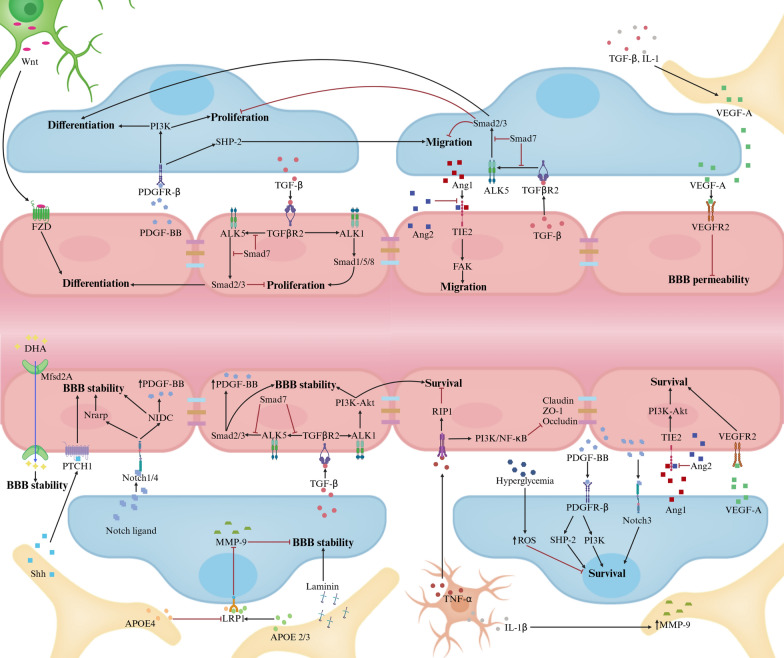
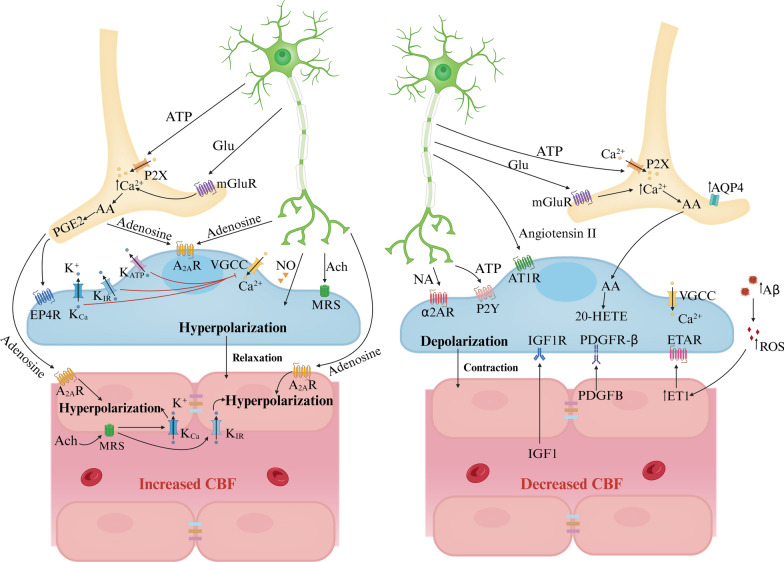
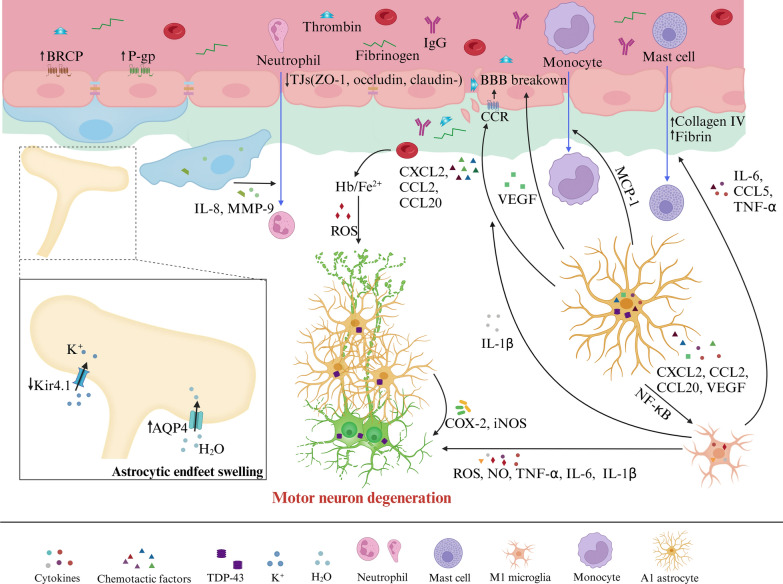
Main body: Early indicators of multiple neurodegenerative disorders in humans and animal models include impaired BBB stability, regional cerebral blood flow shortfalls, and vascular inflammation associated with BBB dysfunction. Understanding the cellular and molecular mechanisms of BBB dysfunction in brain disorders is crucial for elucidating the sustenance of neural computations under pathological conditions and for developing treatments for these diseases. This paper initially explores the cellular and molecular definition of the BBB, along with the signaling pathways regulating BBB stability, cerebral blood flow, and vascular inflammation. Subsequently, we review current insights into BBB dynamics in Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, and multiple sclerosis. The paper concludes by proposing a unified mechanism whereby BBB dysfunction contributes to neurodegenerative disorders, highlights potential BBB-focused therapeutic strategies and targets, and outlines lessons learned and future research directions.
Conclusions: BBB breakdown significantly impacts the development and progression of neurodegenerative diseases, and unraveling the cellular and molecular mechanisms underlying BBB dysfunction is vital to elucidate how neural computations are sustained under pathological conditions and to devise therapeutic approaches.
Keywords: Blood–brain barrier; Cerebrovascular blood flow; Neurodegenerative diseases; Therapeutics; Vascular inflammation.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Grants and funding
- LGD21H250001 and LGD22H250001/Basic Public Welfare Research Program of Zhejiang Province
- LGD21H250001 and LGD22H250001/Basic Public Welfare Research Program of Zhejiang Province
- LGD21H250001 and LGD22H250001/Basic Public Welfare Research Program of Zhejiang Province
- LGD21H250001 and LGD22H250001/Basic Public Welfare Research Program of Zhejiang Province
- LGD21H250001 and LGD22H250001/Basic Public Welfare Research Program of Zhejiang Province
- LGD21H250001 and LGD22H250001/Basic Public Welfare Research Program of Zhejiang Province
- LGD21H250001 and LGD22H250001/Basic Public Welfare Research Program of Zhejiang Province
- LGD21H250001 and LGD22H250001/Basic Public Welfare Research Program of Zhejiang Province
- LGD21H250001 and LGD22H250001/Basic Public Welfare Research Program of Zhejiang Province
- No.X-202102/Scientific Research Foundation of Zhejiang University City College
LinkOut - more resources
Full Text Sources
Medical
