

ER calcium depletion as a key driver for impaired ER-to-mitochondria calcium transfer and mitochondrial dysfunction in Wolfram syndrome
- PMID: 39034309
- PMCID: PMC11271478
- DOI: 10.1038/s41467-024-50502-x
ER calcium depletion as a key driver for impaired ER-to-mitochondria calcium transfer and mitochondrial dysfunction in Wolfram syndrome
Abstract
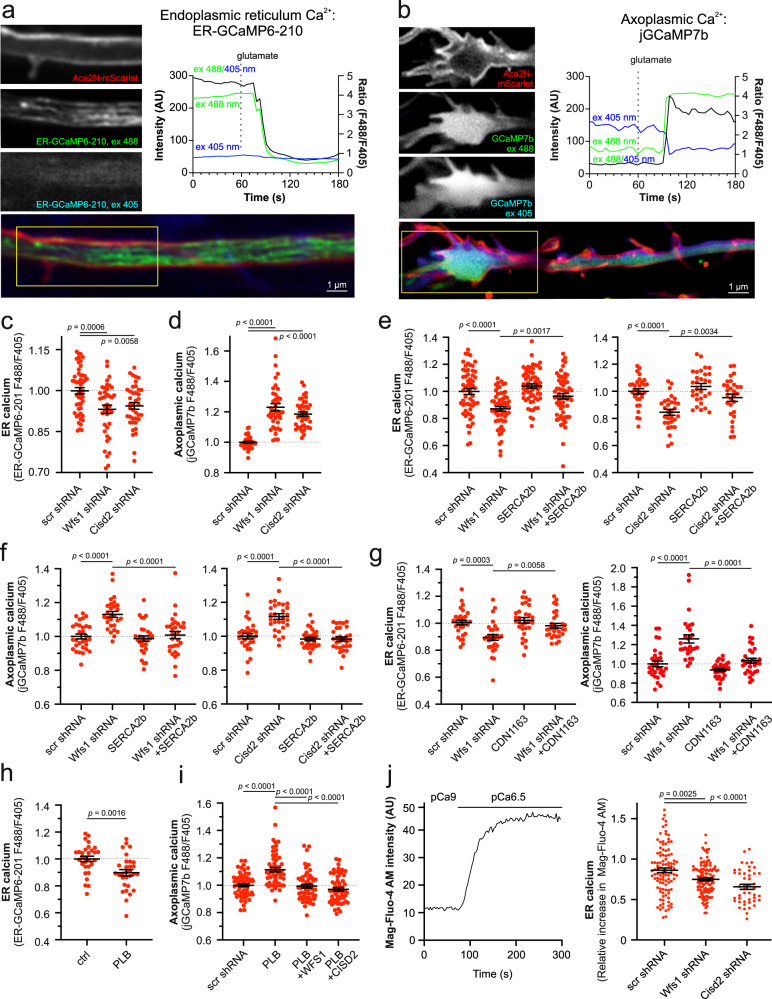
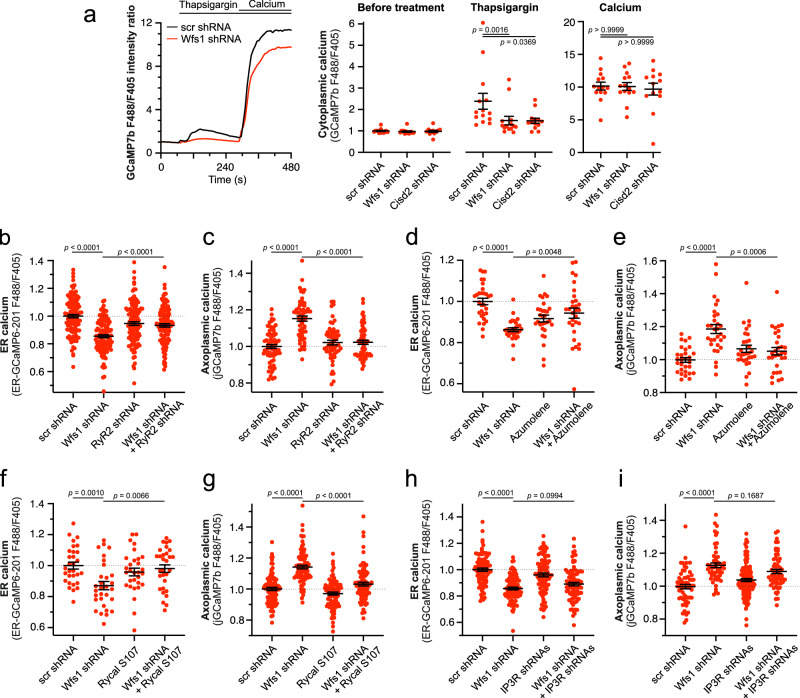
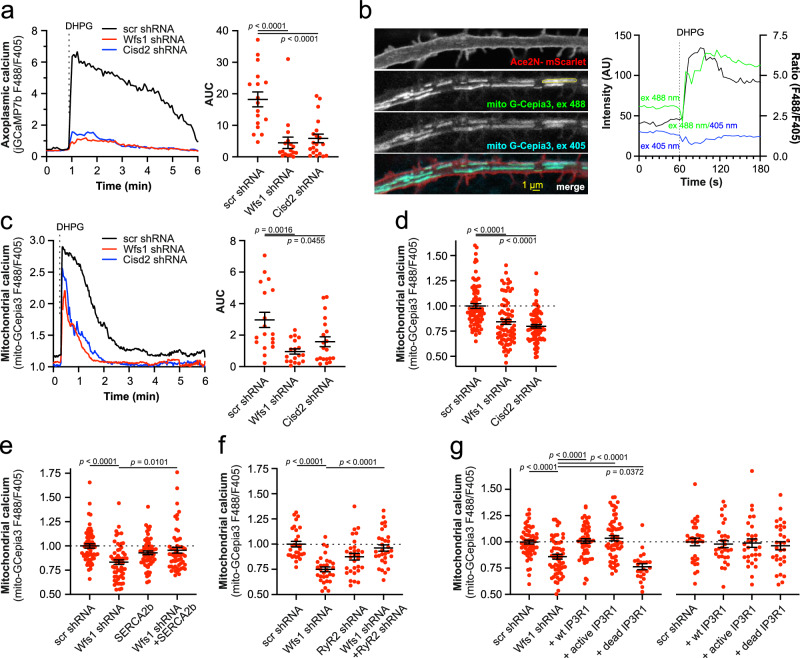
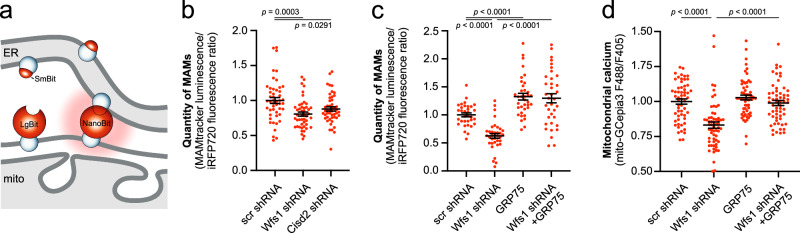
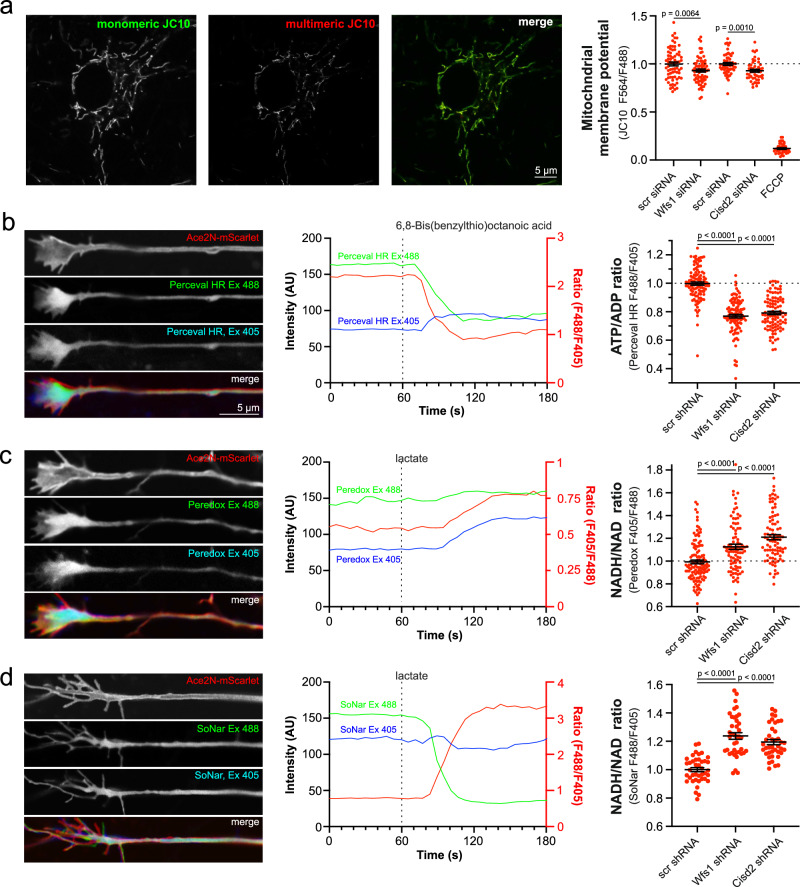
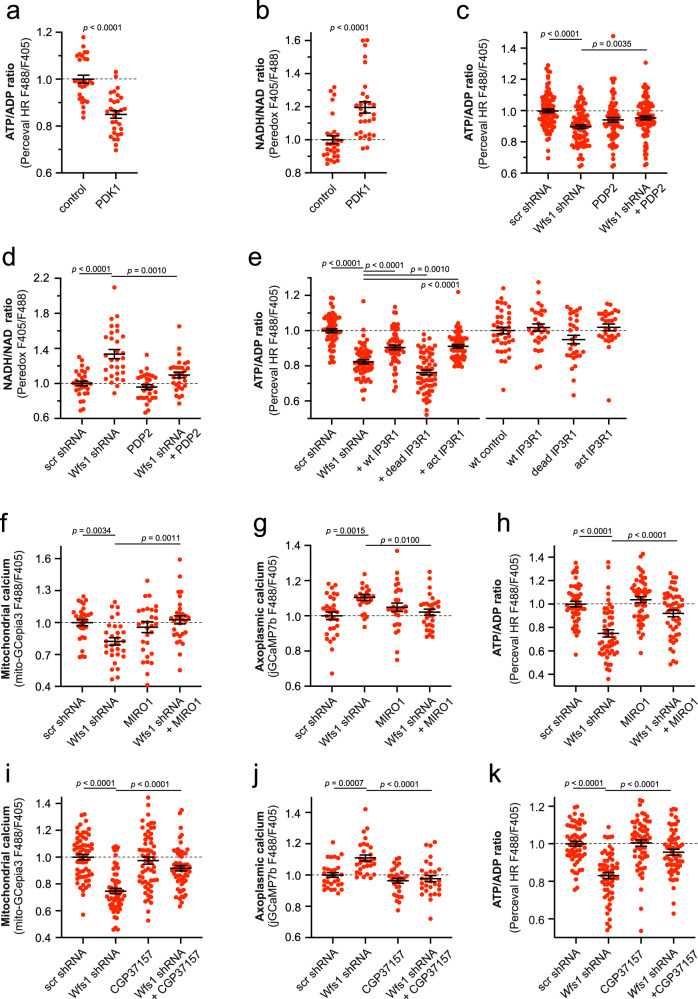
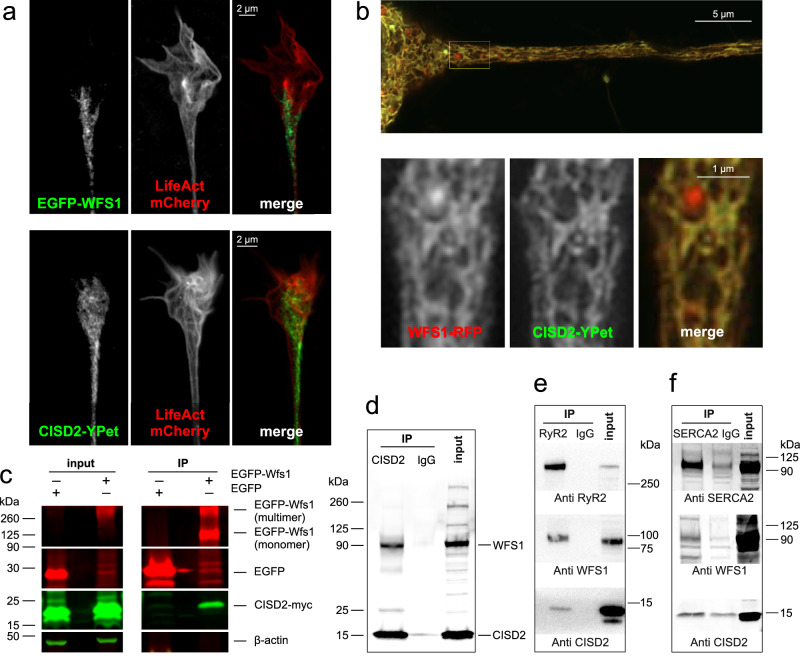
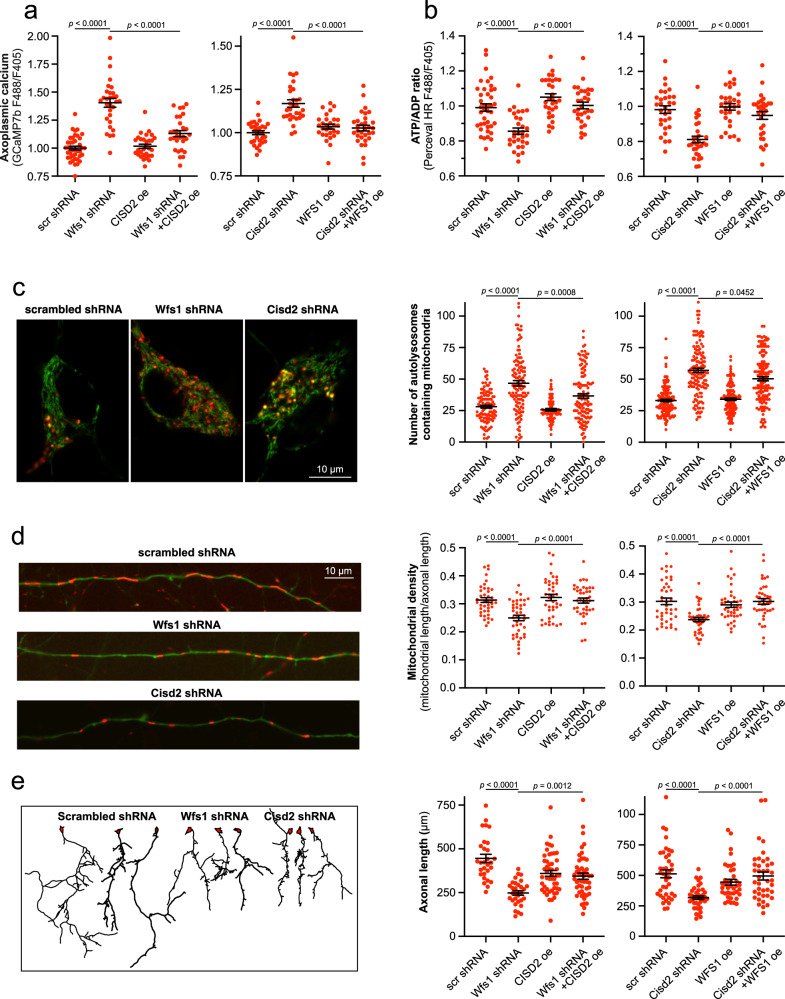
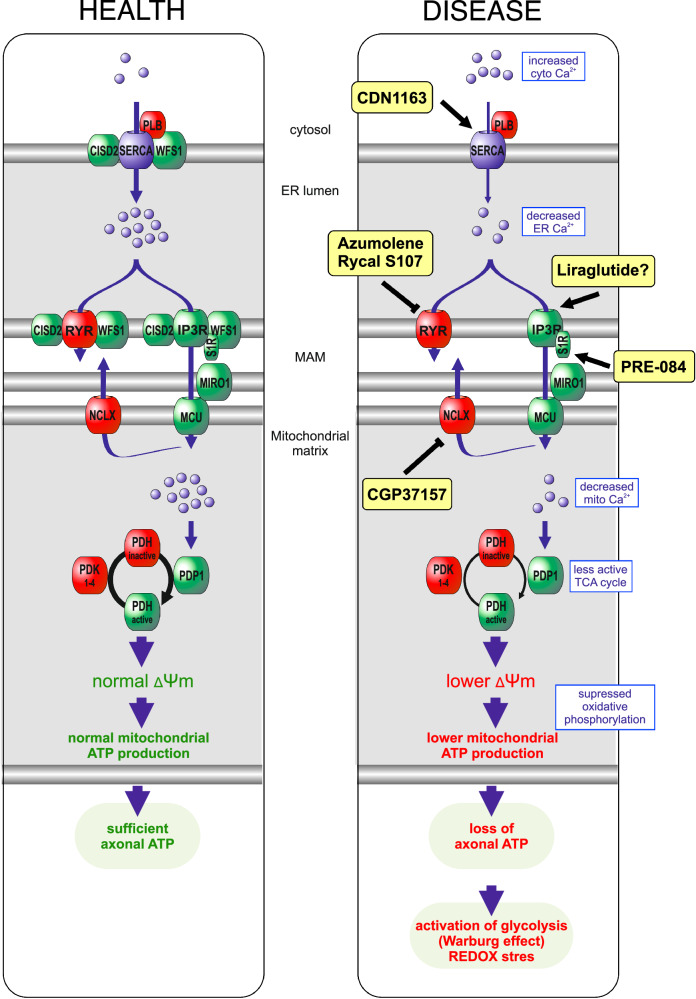
Wolfram syndrome is a rare genetic disease caused by mutations in the WFS1 or CISD2 gene. A primary defect in Wolfram syndrome involves poor ER Ca2+ handling, but how this disturbance leads to the disease is not known. The current study, performed in primary neurons, the most affected and disease-relevant cells, involving both Wolfram syndrome genes, explains how the disturbed ER Ca2+ handling compromises mitochondrial function and affects neuronal health. Loss of ER Ca2+ content and impaired ER-mitochondrial contact sites in the WFS1- or CISD2-deficient neurons is associated with lower IP3R-mediated Ca2+ transfer from ER to mitochondria and decreased mitochondrial Ca2+ uptake. In turn, reduced mitochondrial Ca2+ content inhibits mitochondrial ATP production leading to an increased NADH/NAD+ ratio. The resulting bioenergetic deficit and reductive stress compromise the health of the neurons. Our work also identifies pharmacological targets and compounds that restore Ca2+ homeostasis, enhance mitochondrial function and improve neuronal health.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Barrett, T. et al. In GeneReviews(®) (eds. M. P. Adam, D. B. Everman, & G. M. Mirzaa et al.) (University of Washington, Seattle;1993-2022, 1993).
MeSH terms
Substances
Grants and funding
- PRG400/Eesti Teadusagentuur (Estonian Research Council)
- PRG2145/Eesti Teadusagentuur (Estonian Research Council)
- G081821N/Fonds Wetenschappelijk Onderzoek (Research Foundation Flanders)
- G081821N/Fonds Wetenschappelijk Onderzoek (Research Foundation Flanders)
- 313021T081/Slovenská Akadémia Vied (Slovak Academy of Sciences)
LinkOut - more resources
Full Text Sources
Miscellaneous
