

Quantifying Induced Dipole Effects in Small Molecule Permeation in a Model Phospholipid Bilayer
- PMID: 39038441
- PMCID: PMC11301690
- DOI: 10.1021/acs.jpcb.4c01634
Quantifying Induced Dipole Effects in Small Molecule Permeation in a Model Phospholipid Bilayer
Abstract
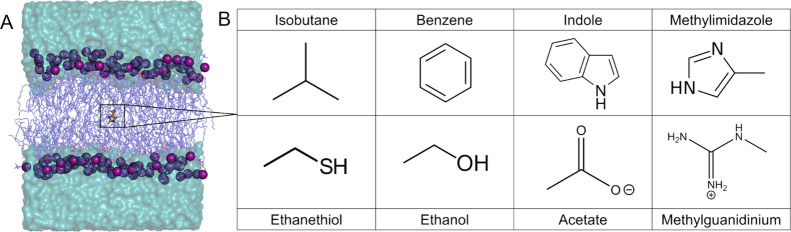
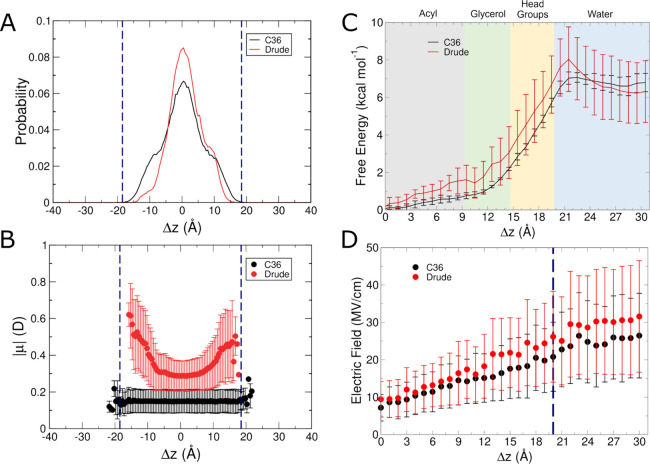
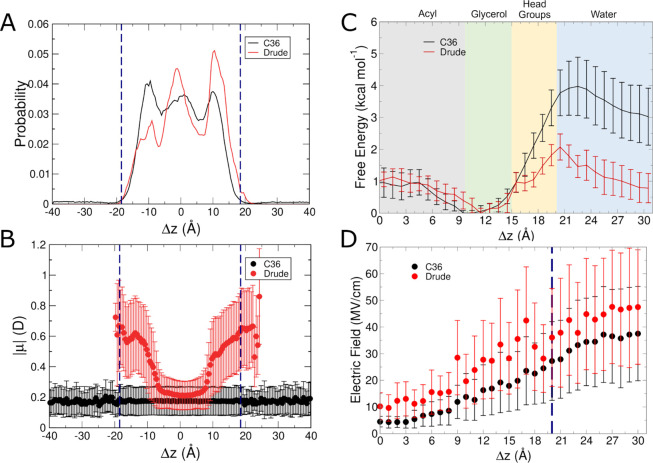
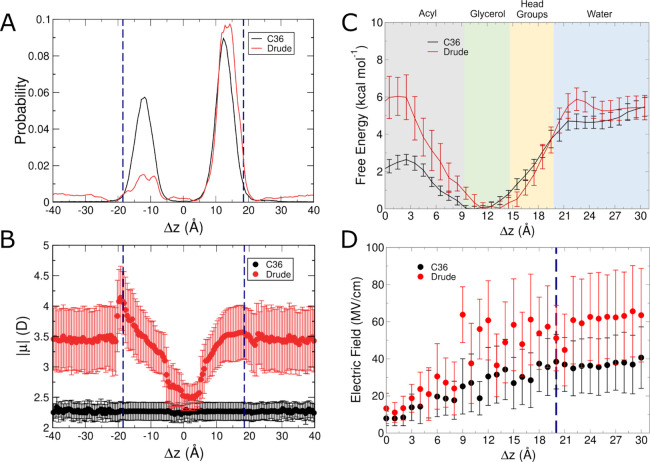
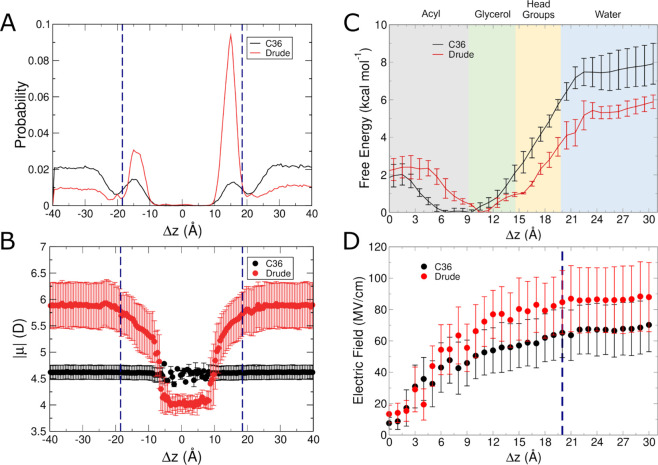
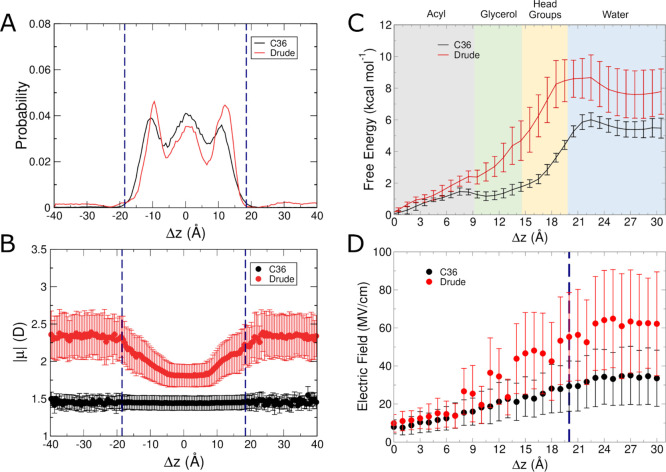
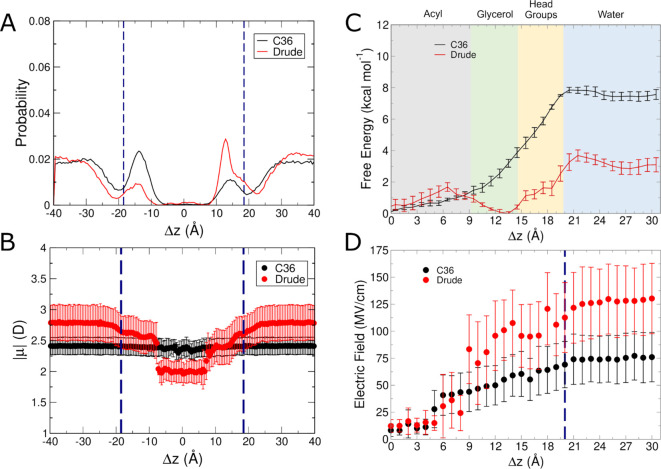
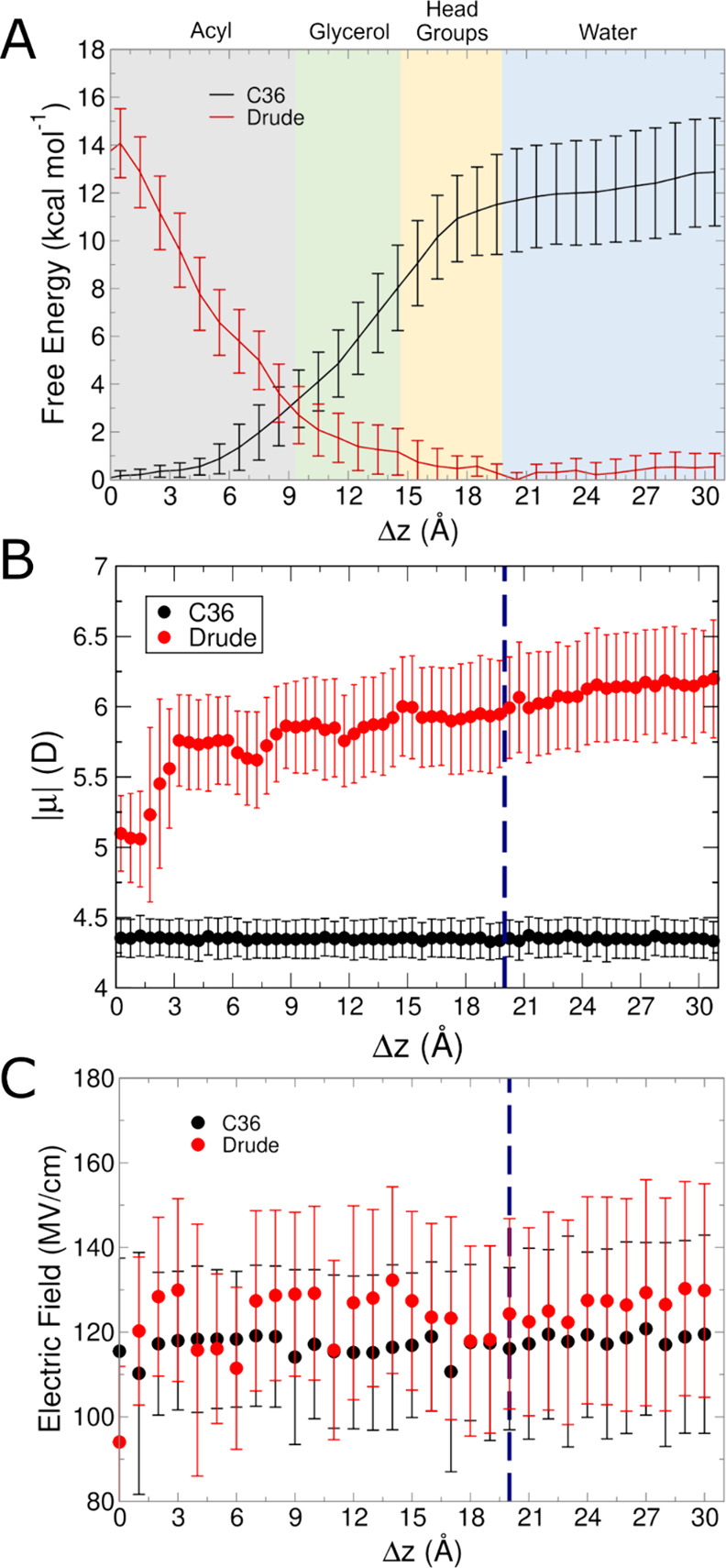
The cell membrane functions as a semipermeable barrier that governs the transport of materials into and out of cells. The bilayer features a distinct dielectric gradient due to the amphiphilic nature of its lipid components. This gradient influences various aspects of small molecule permeation and the folding and functioning of membrane proteins. Here, we employ polarizable molecular dynamics simulations to elucidate the impact of the electronic environment on the permeation process. We simulated eight distinct amino-acid side chain analogs within a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine bilayer using the Drude polarizable force field (FF). Our approach includes both unbiased and umbrella sampling simulations. By using a polarizable FF, we sought to investigate explicit dipole responses in relation to local electric fields along the membrane normal. We evaluate molecular dipole moments, which exhibit variation based on their localization within the membrane, and compare the outcomes with analogous simulations using the nonpolarizable CHARMM36 FF. This comparative analysis aims to discern characteristic differences in the free energy surfaces of permeation for the various amino-acid analogs. Our results provide the first systematic quantification of the impact of employing an explicitly polarizable FF in this context compared to the fixed-charge convention inherent to nonpolarizable FFs, which may not fully capture the influence of the membrane dielectric gradient.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
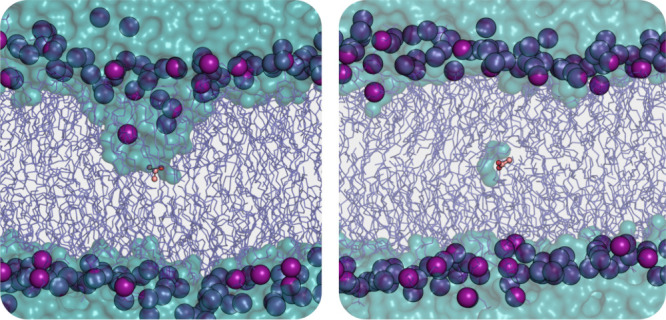
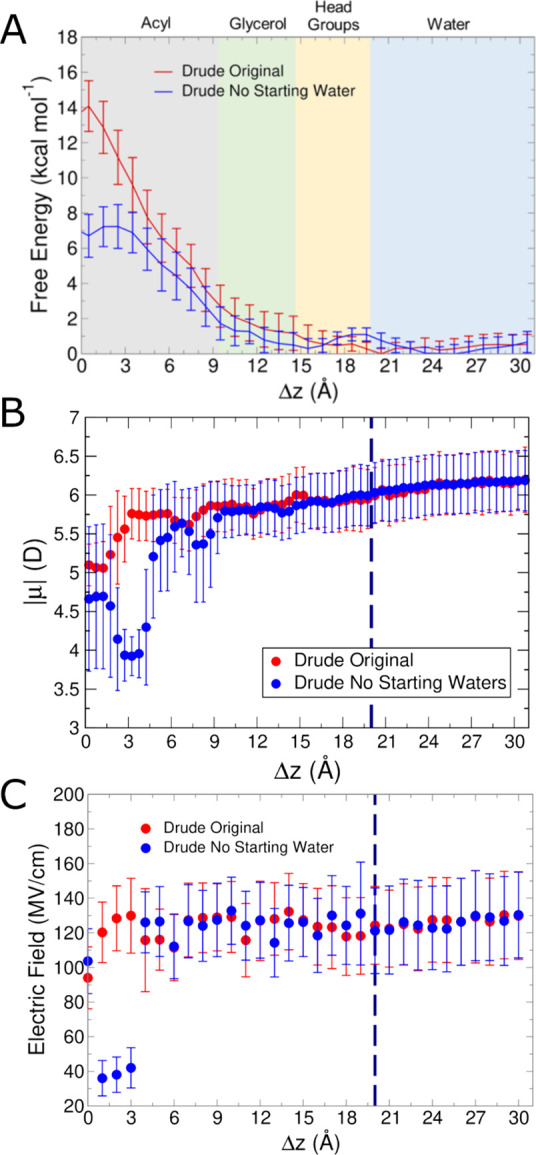
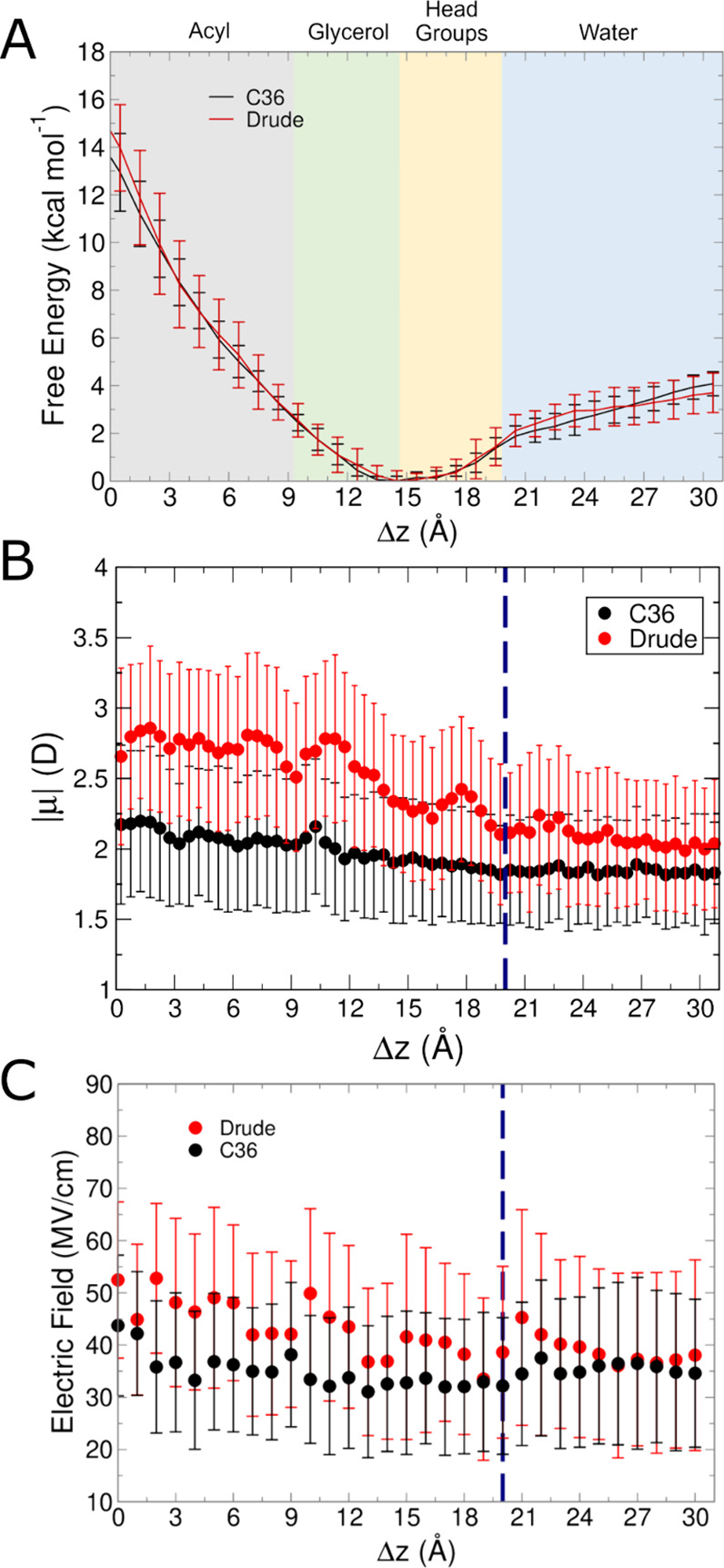
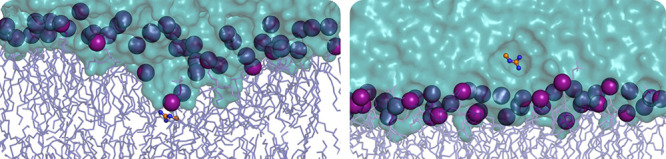
Similar articles
-
Drude Polarizable Force Field for Molecular Dynamics Simulations of Saturated and Unsaturated Zwitterionic Lipids.J Chem Theory Comput. 2017 Sep 12;13(9):4535-4552. doi: 10.1021/acs.jctc.7b00262. Epub 2017 Aug 8. J Chem Theory Comput. 2017. PMID: 28731702 Free PMC article.
-
Molecular dynamics simulations of a DMPC bilayer using nonadditive interaction models.Biophys J. 2009 Jan;96(2):385-402. doi: 10.1016/j.bpj.2008.09.048. Biophys J. 2009. PMID: 19167291 Free PMC article.
-
Polarizable atomic multipole-based force field for cholesterol.J Biomol Struct Dyn. 2024 Sep;42(15):7747-7757. doi: 10.1080/07391102.2023.2245045. Epub 2023 Aug 11. J Biomol Struct Dyn. 2024. PMID: 37565356
-
Unassisted N-acetyl-phenylalanine-amide transport across membrane with varying lipid size and composition: kinetic measurements and atomistic molecular dynamics simulation.J Biomol Struct Dyn. 2022 Mar;40(4):1445-1460. doi: 10.1080/07391102.2020.1827037. Epub 2020 Oct 9. J Biomol Struct Dyn. 2022. PMID: 33034537
-
Charge equilibration force fields for molecular dynamics simulations of lipids, bilayers, and integral membrane protein systems.Biochim Biophys Acta. 2012 Feb;1818(2):318-29. doi: 10.1016/j.bbamem.2011.09.016. Epub 2011 Sep 24. Biochim Biophys Acta. 2012. PMID: 21967961 Free PMC article. Review.
References
-
- Egberts E.; Berendsen H. J. C. Molecular Dynamics Simulation of a Smectic Liquid Crystal with Atomic Detail. J. Chem. Phys. 1988, 89 (6), 3718–3732. 10.1063/1.454893. - DOI
-
- Jönsson B.; Edholm O.; Teleman O. Molecular Dynamics Simulations of a Sodium Octanoate Micelle in Aqueous Solution. J. Chem. Phys. 1986, 85 (4), 2259–2271. 10.1063/1.451122. - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
