

Next-generation phenotyping integrated in a national framework for patients with ultrarare disorders improves genetic diagnostics and yields new molecular findings
- PMID: 39039281
- PMCID: PMC11319204
- DOI: 10.1038/s41588-024-01836-1
Next-generation phenotyping integrated in a national framework for patients with ultrarare disorders improves genetic diagnostics and yields new molecular findings
Erratum in
-
Author Correction: Next-generation phenotyping integrated in a national framework for patients with ultrarare disorders improves genetic diagnostics and yields new molecular findings.Nat Genet. 2025 Jul;57(7):1790-1791. doi: 10.1038/s41588-025-02271-6. Nat Genet. 2025. PMID: 40555819 Free PMC article. No abstract available.
Abstract
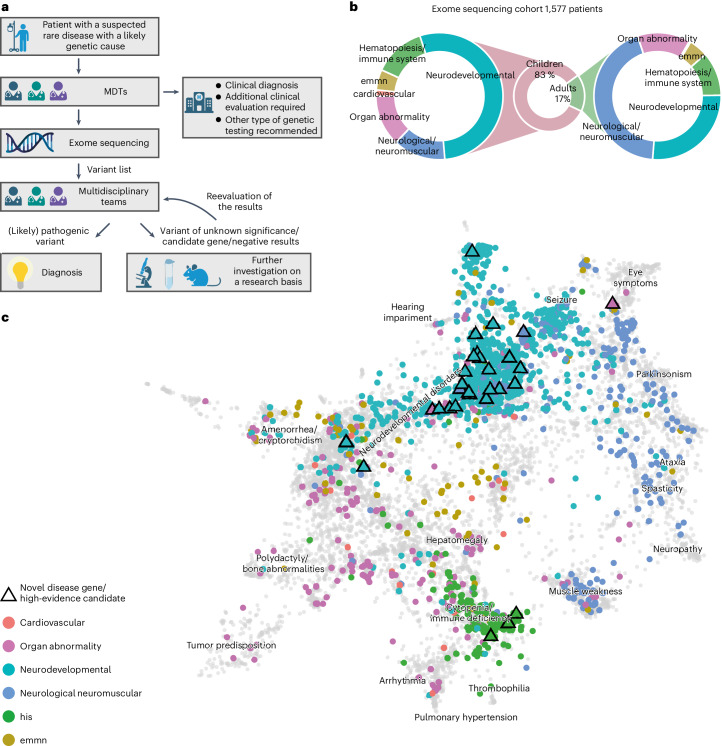
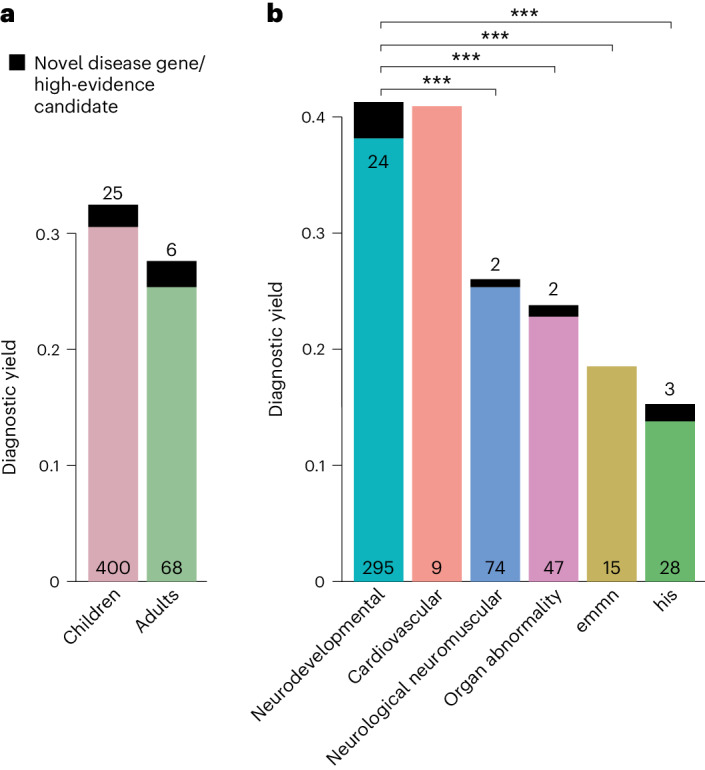
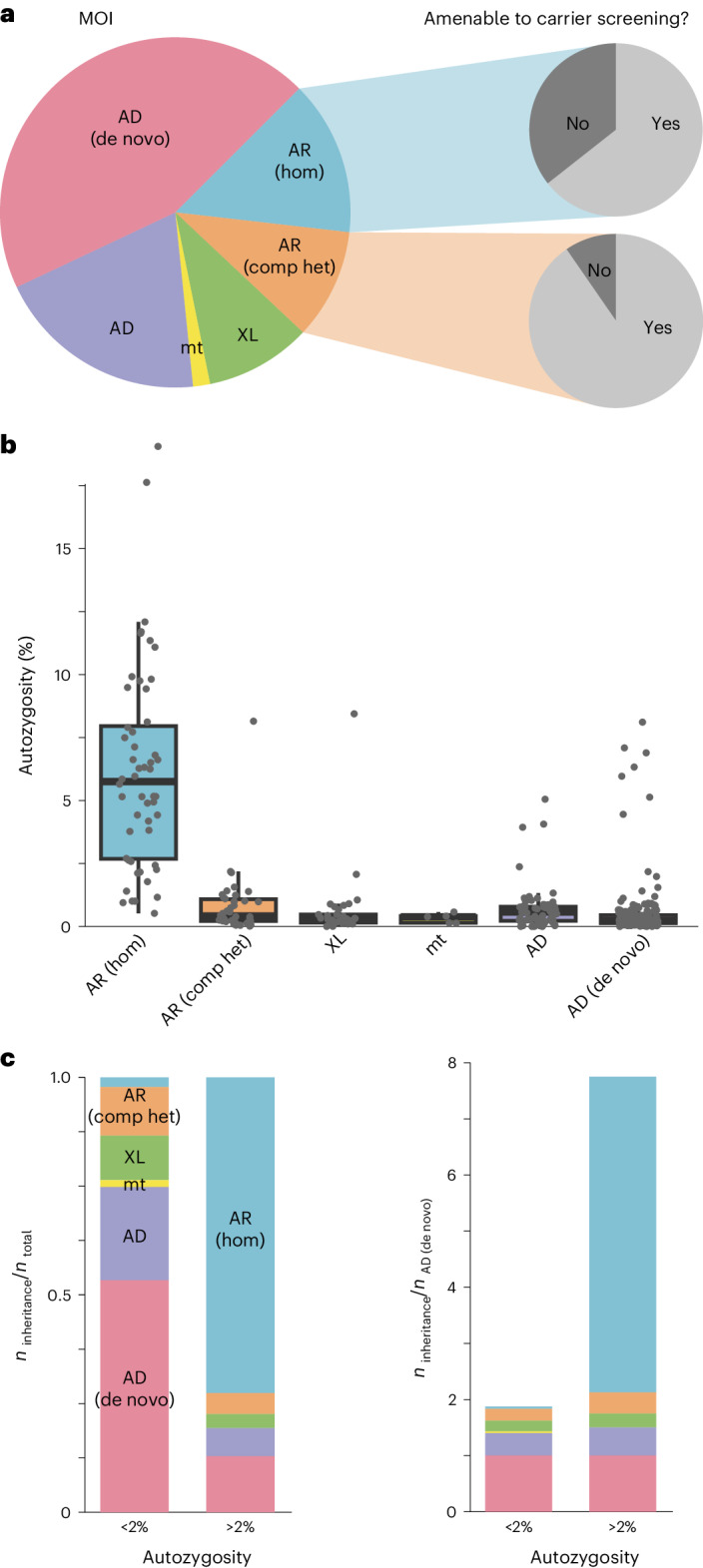
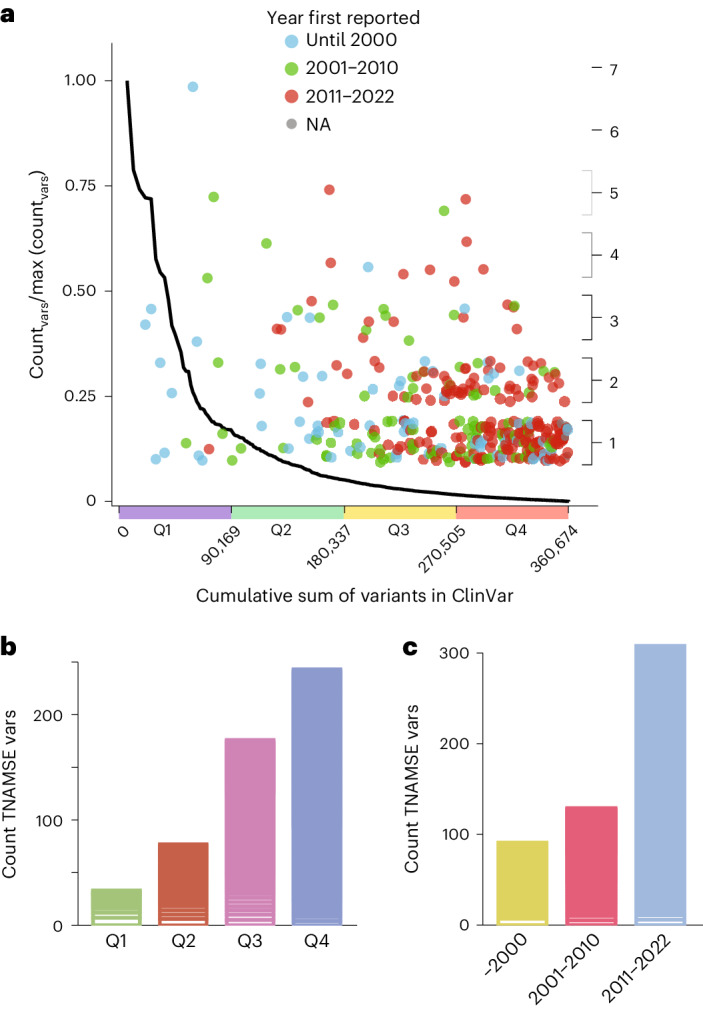
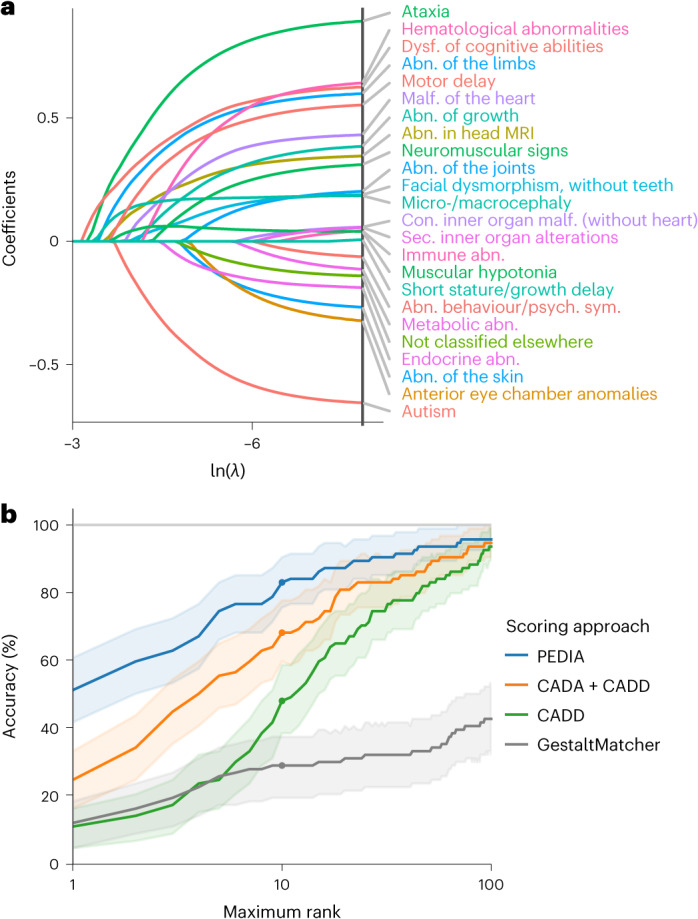
Individuals with ultrarare disorders pose a structural challenge for healthcare systems since expert clinical knowledge is required to establish diagnoses. In TRANSLATE NAMSE, a 3-year prospective study, we evaluated a novel diagnostic concept based on multidisciplinary expertise in Germany. Here we present the systematic investigation of the phenotypic and molecular genetic data of 1,577 patients who had undergone exome sequencing and were partially analyzed with next-generation phenotyping approaches. Molecular genetic diagnoses were established in 32% of the patients totaling 370 distinct molecular genetic causes, most with prevalence below 1:50,000. During the diagnostic process, 34 novel and 23 candidate genotype-phenotype associations were identified, mainly in individuals with neurodevelopmental disorders. Sequencing data of the subcohort that consented to computer-assisted analysis of their facial images with GestaltMatcher could be prioritized more efficiently compared with approaches based solely on clinical features and molecular scores. Our study demonstrates the synergy of using next-generation sequencing and phenotyping for diagnosing ultrarare diseases in routine healthcare and discovering novel etiologies by multidisciplinary teams.
© 2024. The Author(s).
Conflict of interest statement
V.S.S. has received consultant fees from Novartis, Chugai, AbbVie, Celgene, Sanofi, Lilly, Hexal, Pfizer, Amgen, BMS, Roche, Gilead, Medac, Boehringer-Ingelheim and Alexion and speaker’s bureau fees from AbbVie, Novartis, BMS, Chugai, Celgene, Medac, Sanofi, Lilly, Hexal, Pfizer, Janssen, Roche, Schire, Onkowissen, Royal College London, Boehringer-Ingelheim and UCB Fresenius. M.G.-E. has received research support from the German Ministry of Education and Research (BMBF) within the European Joint Program for Rare Diseases (EJP-RD) 2021 Transnational Call for Rare Disease Research Projects (funding number 01GM2110), from the National Ataxia Foundation (NAF) and from Ataxia UK and received consulting fees from Healthcare Manufaktur, Germany, all unrelated to this study. All other authors declare no competing interests.
Figures
References
-
- Hochstenbach, R. et al. Array analysis and karyotyping: workflow consequences based on a retrospective study of 36,325 patients with idiopathic developmental delay in the Netherlands. Eur. J. Med. Genet.52, 161–169 (2009). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
