

Unraveling the habitat preferences, ecological drivers, potential hosts, and auxiliary metabolism of soil giant viruses across China
- PMID: 39039586
- PMCID: PMC11265010
- DOI: 10.1186/s40168-024-01851-8
Unraveling the habitat preferences, ecological drivers, potential hosts, and auxiliary metabolism of soil giant viruses across China
Abstract
Background: Soil giant viruses are increasingly believed to have profound effects on ecological functioning by infecting diverse eukaryotes. However, their biogeography and ecology remain poorly understood.
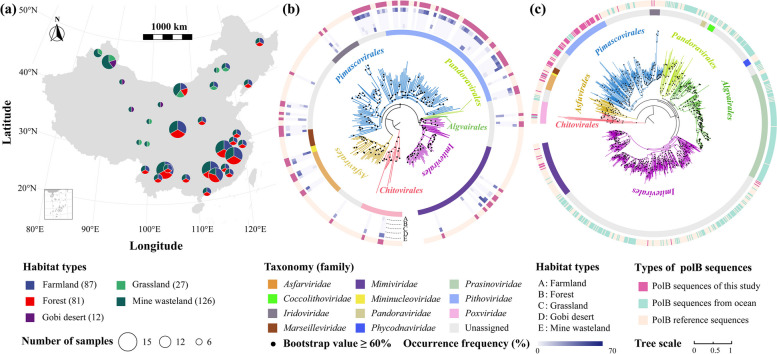
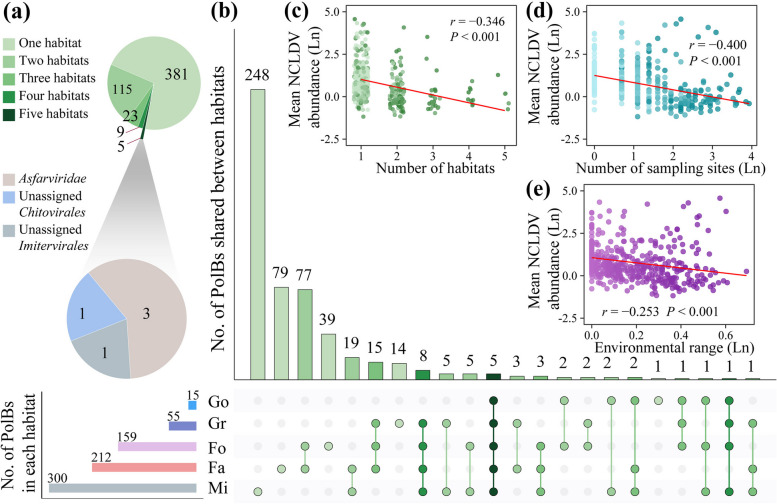
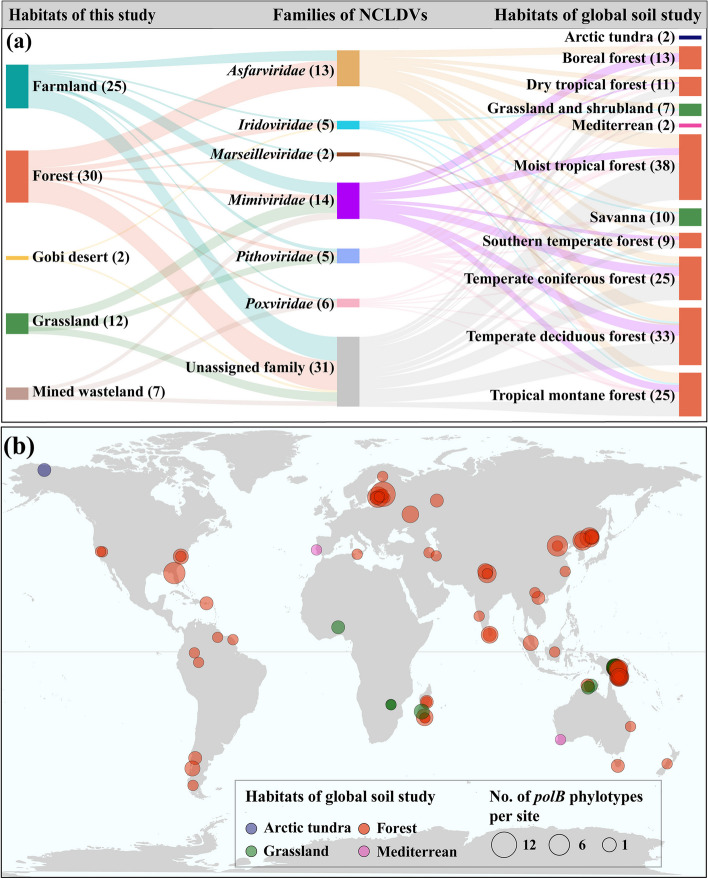
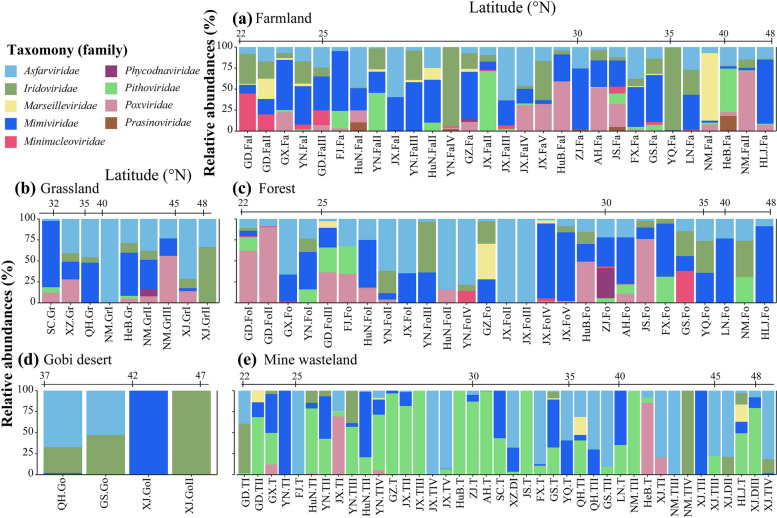
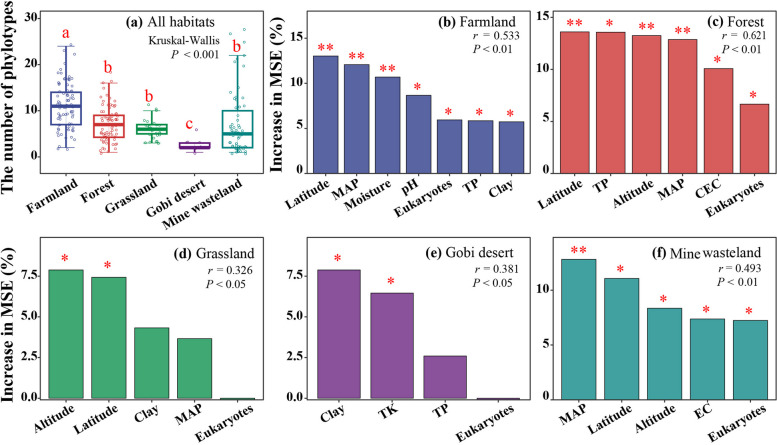
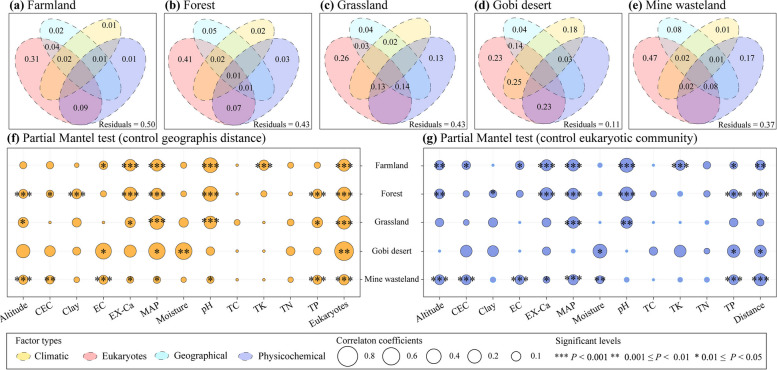
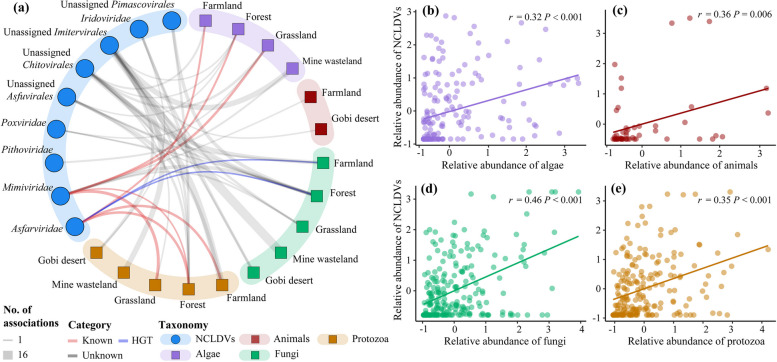
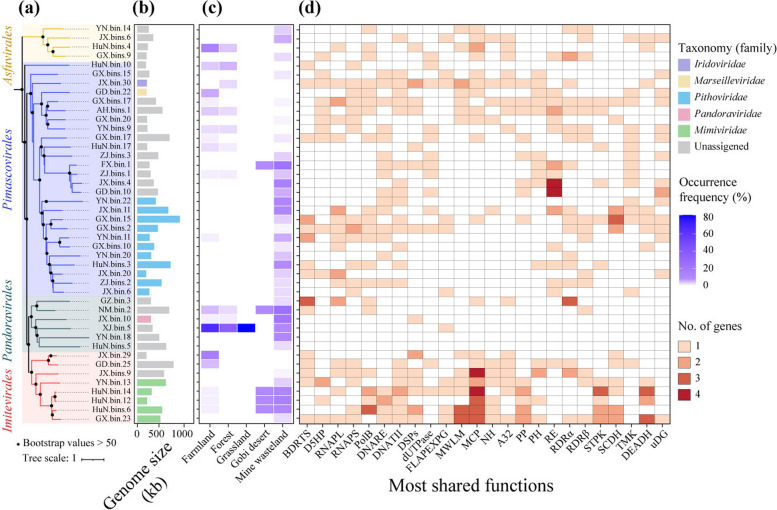
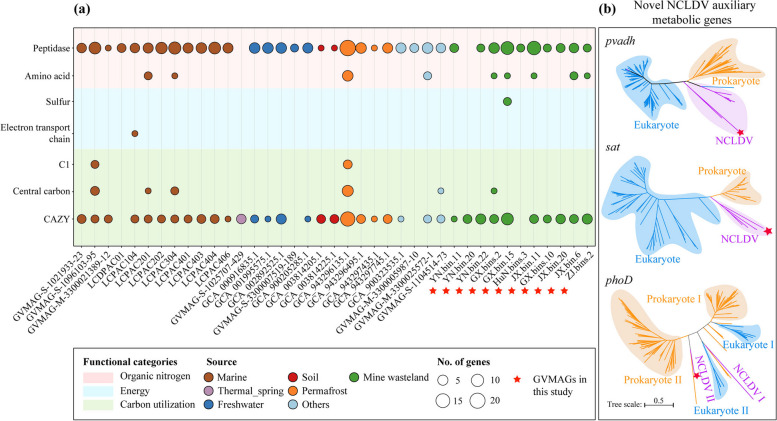
Results: In this study, we analyzed 333 soil metagenomes from 5 habitat types (farmland, forest, grassland, Gobi desert, and mine wasteland) across China and identified 533 distinct giant virus phylotypes affiliated with nine families, thereby greatly expanding the diversity of soil giant viruses. Among the nine families, Pithoviridae were the most diverse. The majority of phylotypes exhibited a heterogeneous distribution among habitat types, with a remarkably high proportion of unique phylotypes in mine wasteland. The abundances of phylotypes were negatively correlated with their environmental ranges. A total of 76 phylotypes recovered in this study were detectable in a published global topsoil metagenome dataset. Among climatic, geographical, edaphic, and biotic characteristics, soil eukaryotes were identified as the most important driver of beta-diversity of giant viral communities across habitat types. Moreover, co-occurrence network analysis revealed some pairings between giant viral phylotypes and eukaryotes (protozoa, fungi, and algae). Analysis of 44 medium- to high-quality giant virus genomes recovered from our metagenomes uncovered not only their highly shared functions but also their novel auxiliary metabolic genes related to carbon, sulfur, and phosphorus cycling.
Conclusions: These findings extend our knowledge of diversity, habitat preferences, ecological drivers, potential hosts, and auxiliary metabolism of soil giant viruses. Video Abstract.
Keywords: Abundance–distribution relationship; Ecological drivers; Eukaryotic community; Geographic distribution; Soil nucleocytoplasmic large DNA viruses; Terrestrial ecosystem.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
