

Multiomic profiling of medulloblastoma reveals subtype-specific targetable alterations at the proteome and N-glycan level
- PMID: 39043693
- PMCID: PMC11266559
- DOI: 10.1038/s41467-024-50554-z
Multiomic profiling of medulloblastoma reveals subtype-specific targetable alterations at the proteome and N-glycan level
Abstract
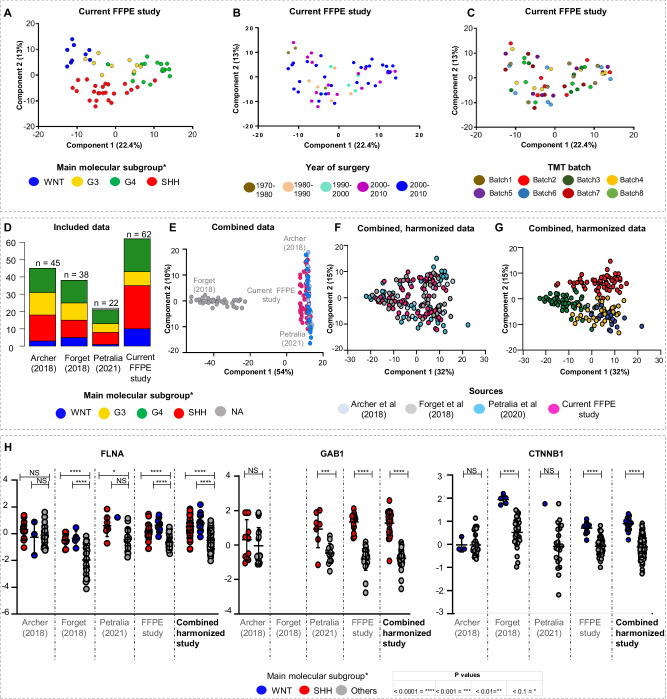
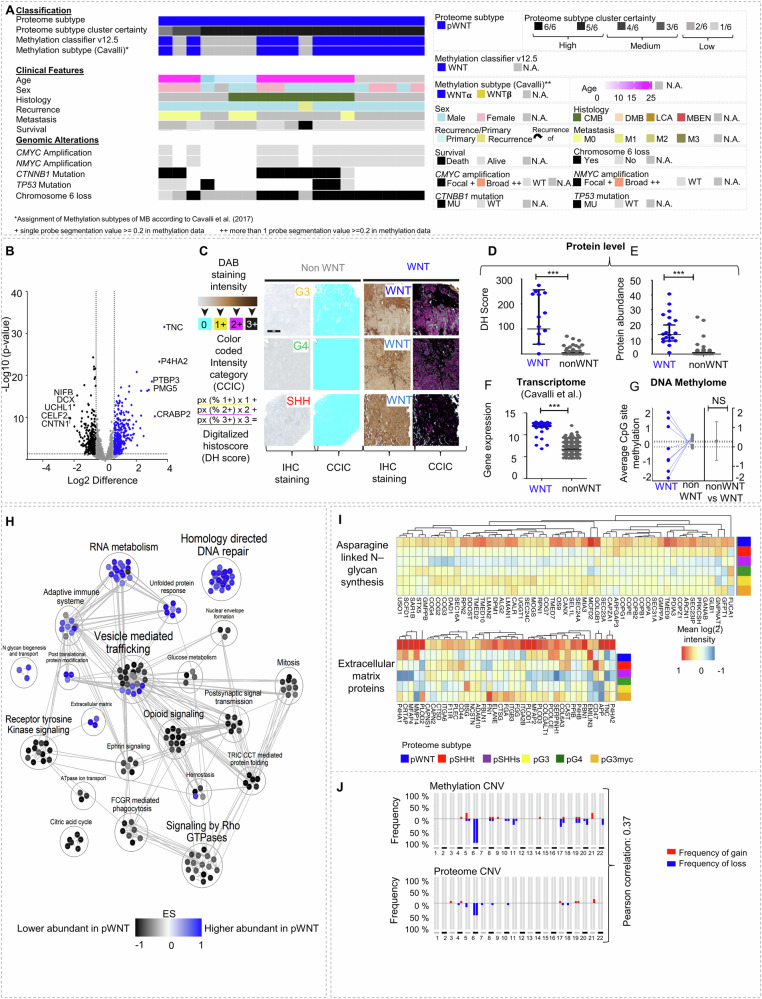
Medulloblastomas (MBs) are malignant pediatric brain tumors that are molecularly and clinically heterogenous. The application of omics technologies-mainly studying nucleic acids-has significantly improved MB classification and stratification, but treatment options are still unsatisfactory. The proteome and their N-glycans hold the potential to discover clinically relevant phenotypes and targetable pathways. We compile a harmonized proteome dataset of 167 MBs and integrate findings with DNA methylome, transcriptome and N-glycome data. We show six proteome MB subtypes, that can be assigned to two main molecular programs: transcription/translation (pSHHt, pWNT and pG3myc), and synapses/immunological processes (pSHHs, pG3 and pG4). Multiomic analysis reveals different conservation levels of proteome features across MB subtypes at the DNA methylome level. Aggressive pGroup3myc MBs and favorable pWNT MBs are most similar in cluster hierarchies concerning overall proteome patterns but show different protein abundances of the vincristine resistance-associated multiprotein complex TriC/CCT and of N-glycan turnover-associated factors. The N-glycome reflects proteome subtypes and complex-bisecting N-glycans characterize pGroup3myc tumors. Our results shed light on targetable alterations in MB and set a foundation for potential immunotherapies targeting glycan structures.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures








References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
