

NMNAT2 is a druggable target to drive neuronal NAD production
- PMID: 39048544
- PMCID: PMC11269627
- DOI: 10.1038/s41467-024-50354-5
NMNAT2 is a druggable target to drive neuronal NAD production
Erratum in
-
Author Correction: NMNAT2 is a druggable target to drive neuronal NAD production.Nat Commun. 2024 Sep 17;15(1):8143. doi: 10.1038/s41467-024-52439-7. Nat Commun. 2024. PMID: 39289370 Free PMC article. No abstract available.
Abstract
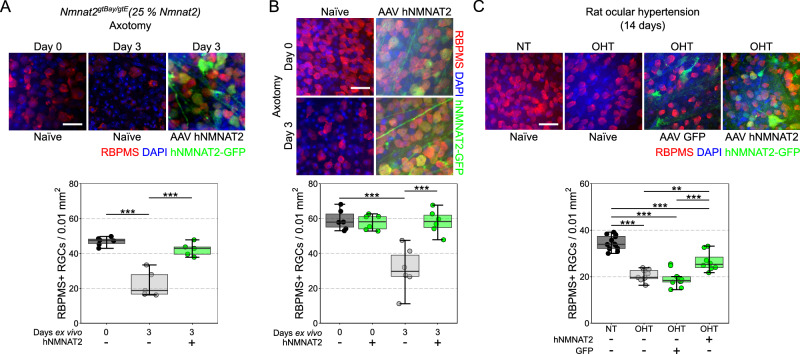
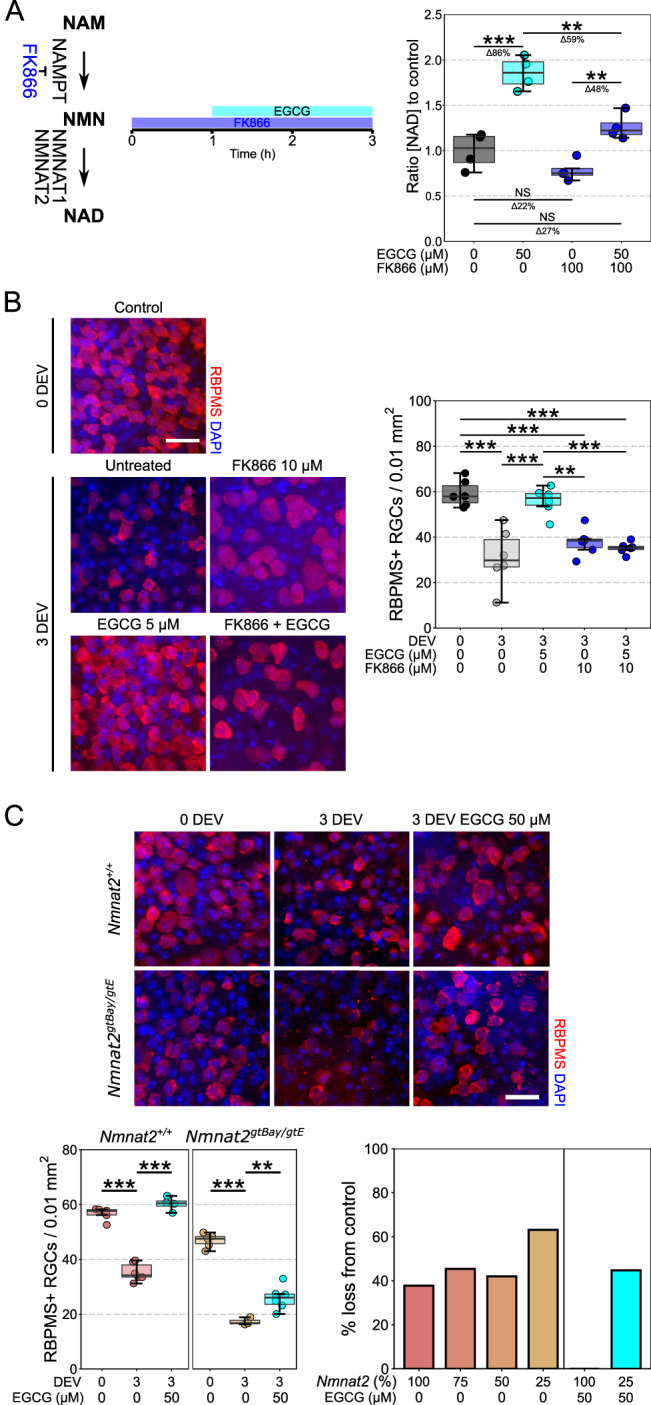
Maintenance of NAD pools is critical for neuronal survival. The capacity to maintain NAD pools declines in neurodegenerative disease. We identify that low NMNAT2, the critical neuronal NAD producing enzyme, drives retinal susceptibility to neurodegenerative insults. As proof of concept, gene therapy over-expressing full length human NMNAT2 is neuroprotective. To pharmacologically target NMNAT2, we identify that epigallocatechin gallate (EGCG) can drive NAD production in neurons through an NMNAT2 and NMN dependent mechanism. We confirm this by pharmacological and genetic inhibition of the NAD-salvage pathway. EGCG is neuroprotective in rodent (mixed sex) and human models of retinal neurodegeneration. As EGCG has poor drug-like qualities, we use it as a tool compound to generate novel small molecules which drive neuronal NAD production and provide neuroprotection. This class of NMNAT2 targeted small molecules could have an important therapeutic impact for neurodegenerative disease following further drug development.
© 2024. The Author(s).
Conflict of interest statement
PAW is an inventor on an awarded US patent held by The Jackson Laboratory for nicotinamide treatment in glaucoma (“Treatment and prevention of ocular neurodegenerative disorder”, US11389439B2). PAW, MJ, CV, and AB are inventors on a submitted patent held by Mim Neurosciences AB for novel NMNAT2-targeting small molecules. All other authors declare that they have no competing interests.
Figures






References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
