

Single-cell multiregion dissection of Alzheimer's disease
- PMID: 39048816
- PMCID: PMC11338834
- DOI: 10.1038/s41586-024-07606-7
Single-cell multiregion dissection of Alzheimer's disease
Abstract
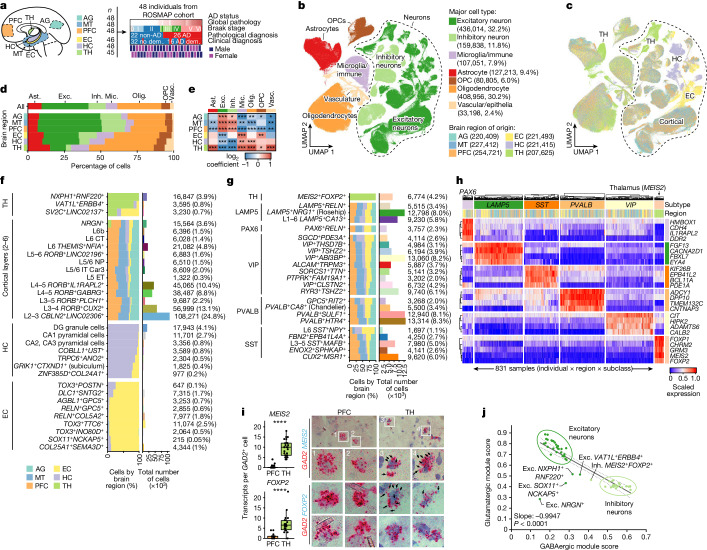
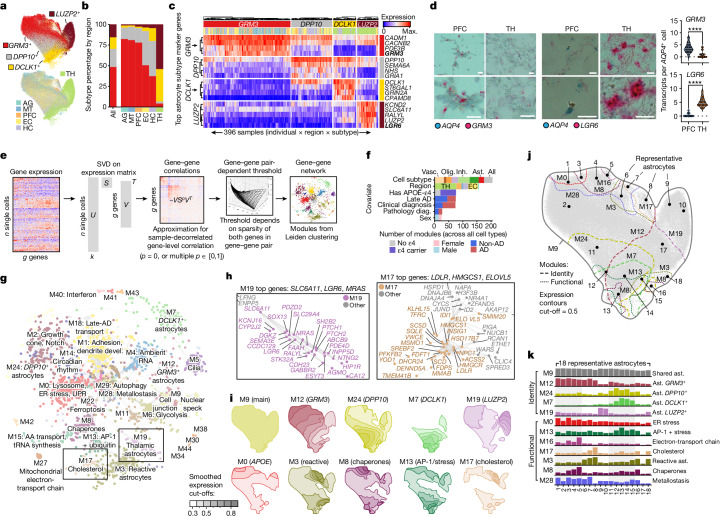
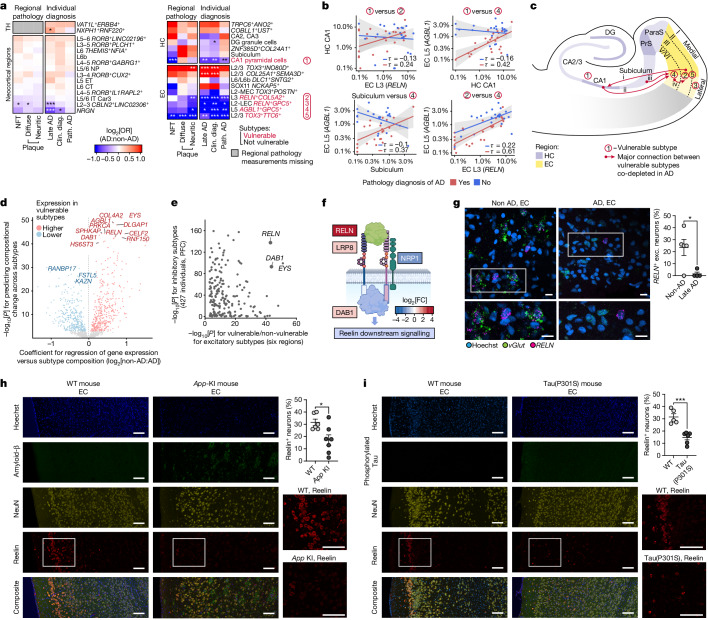
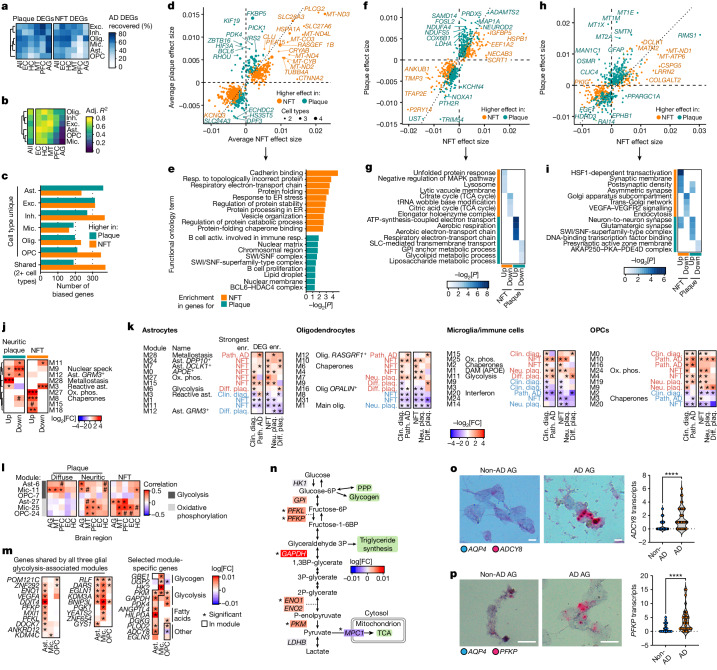
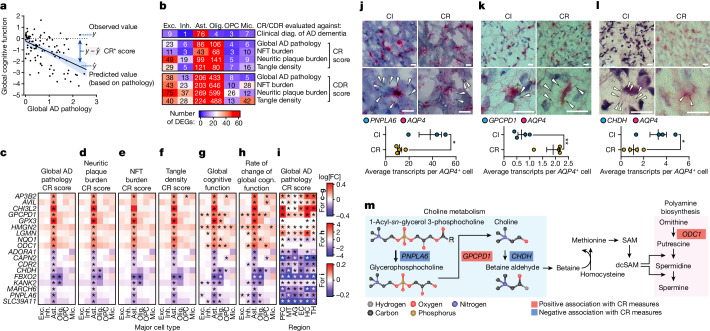
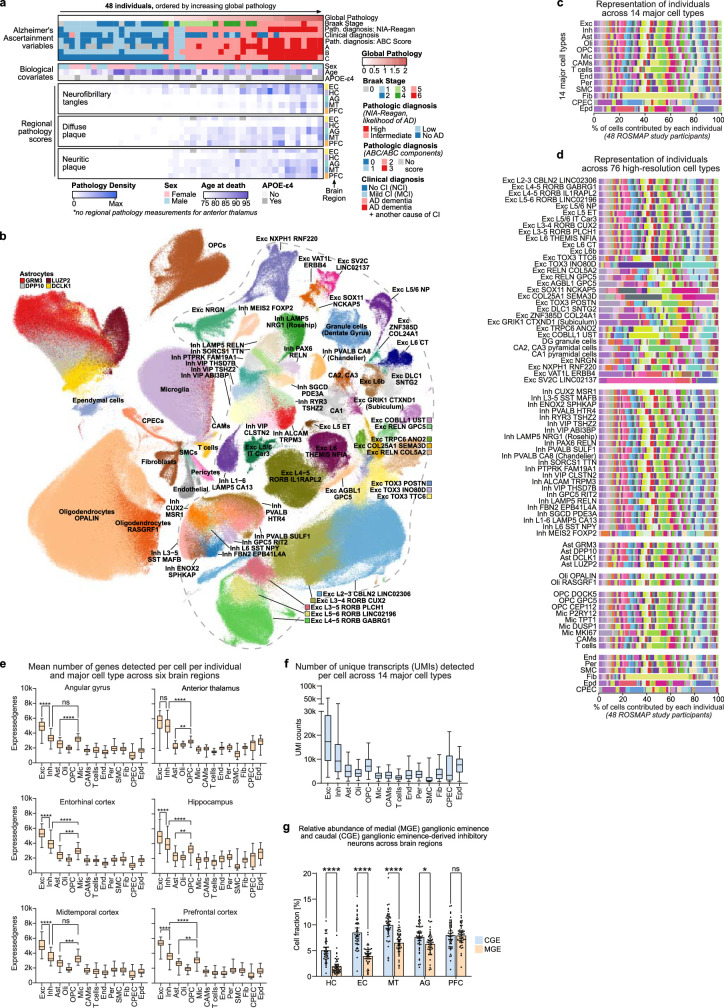
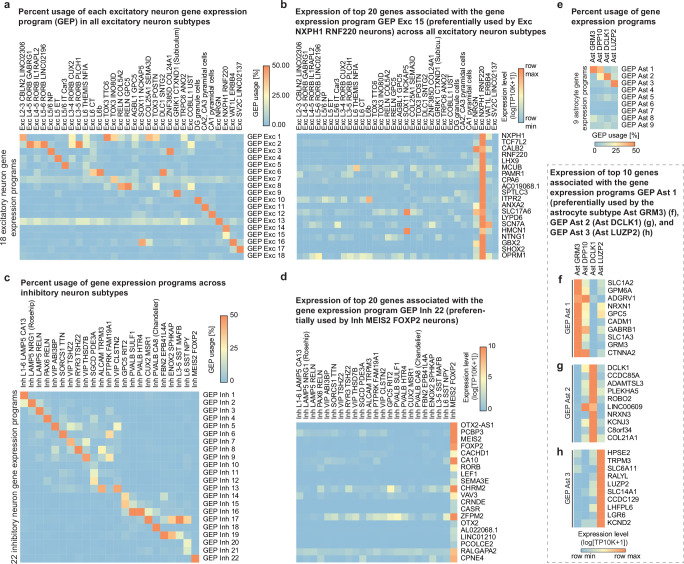
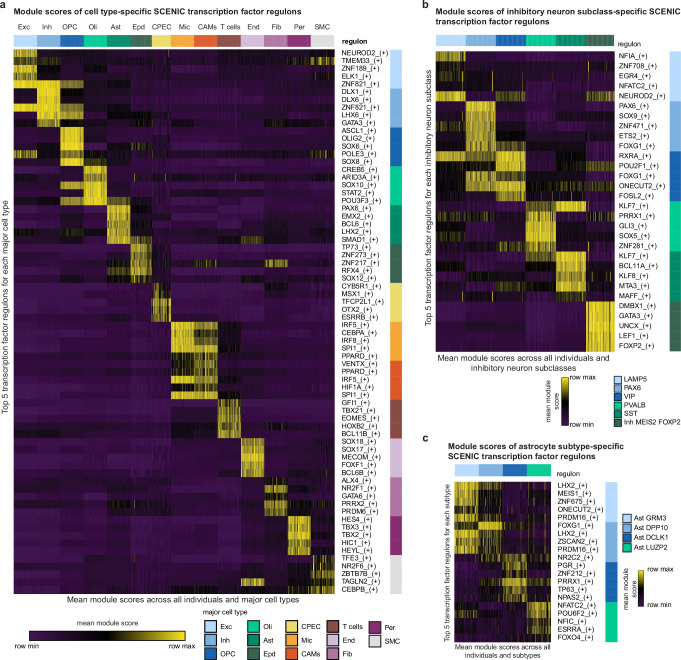
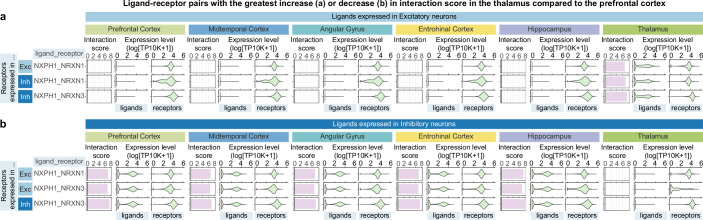
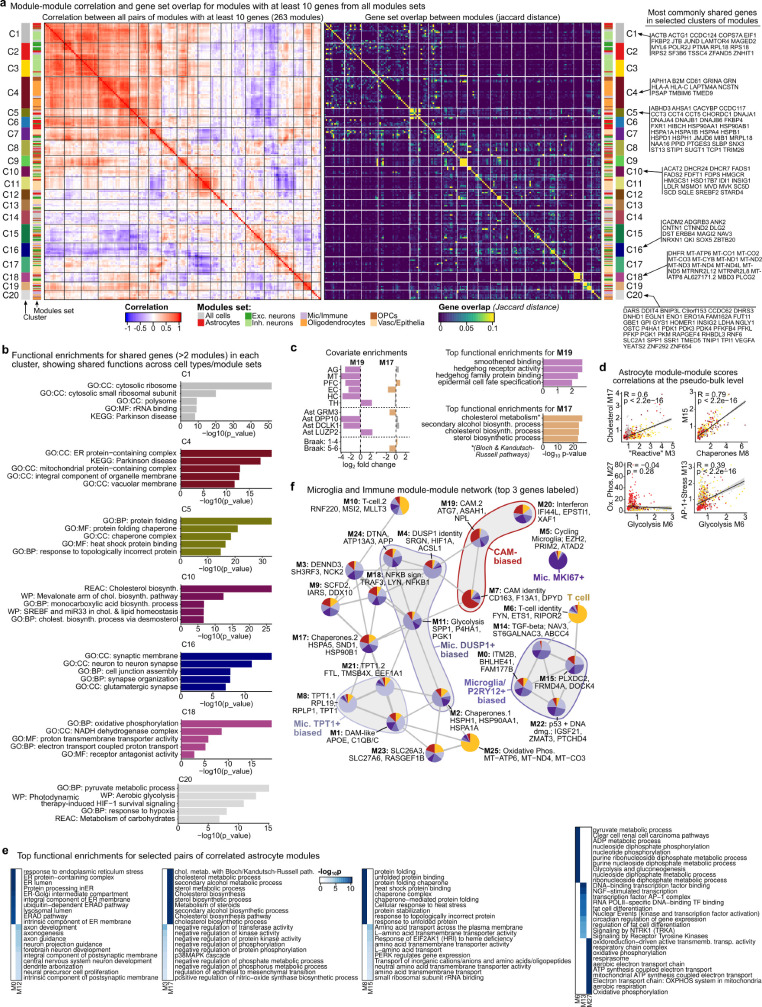
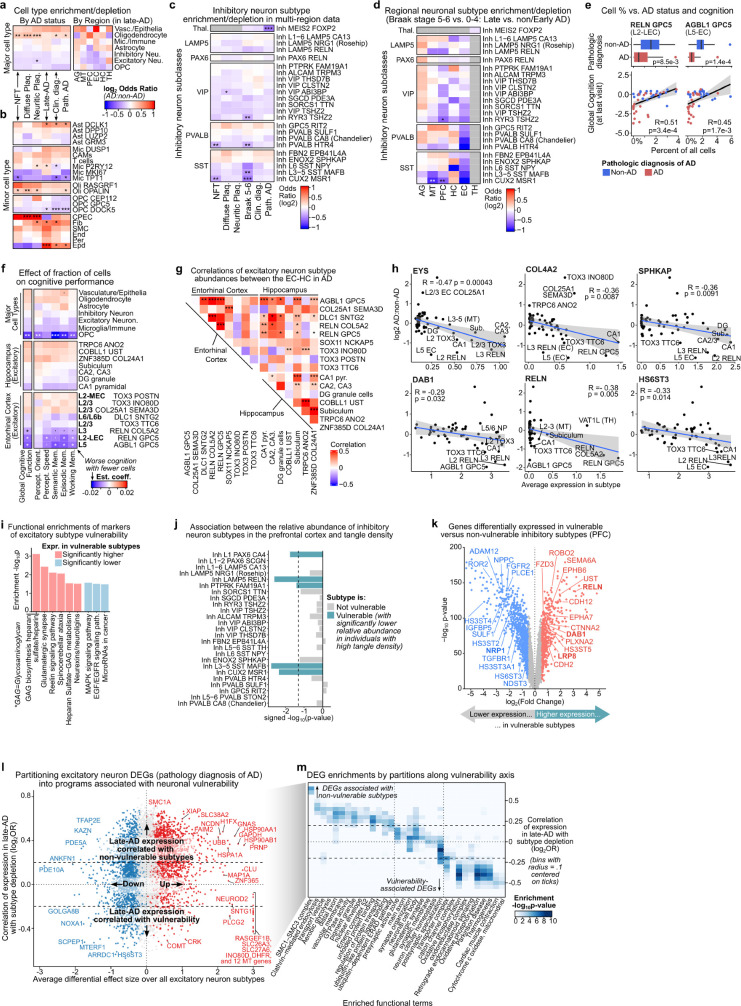
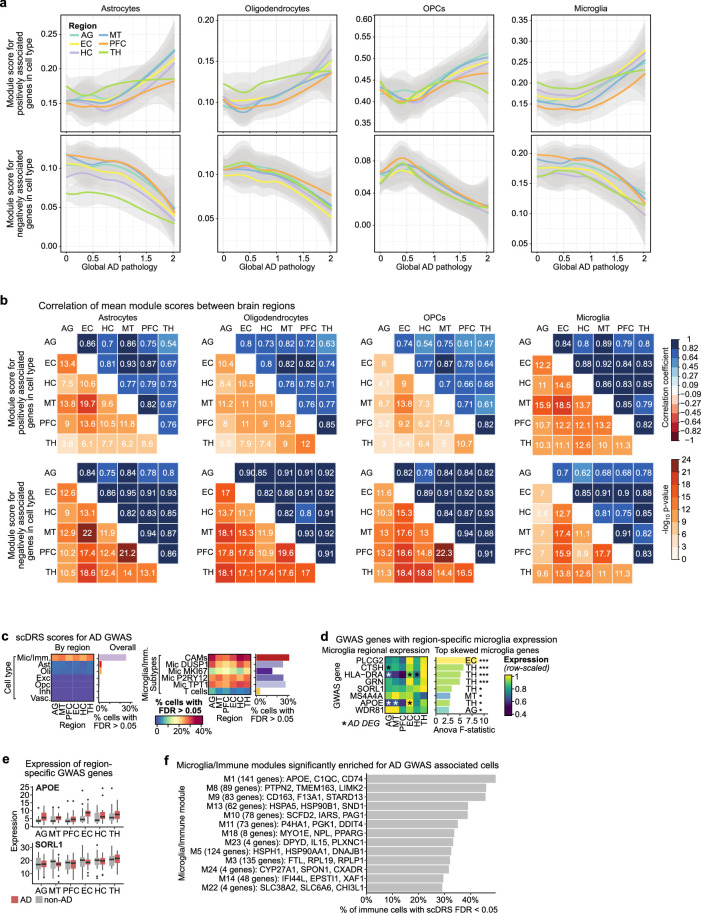
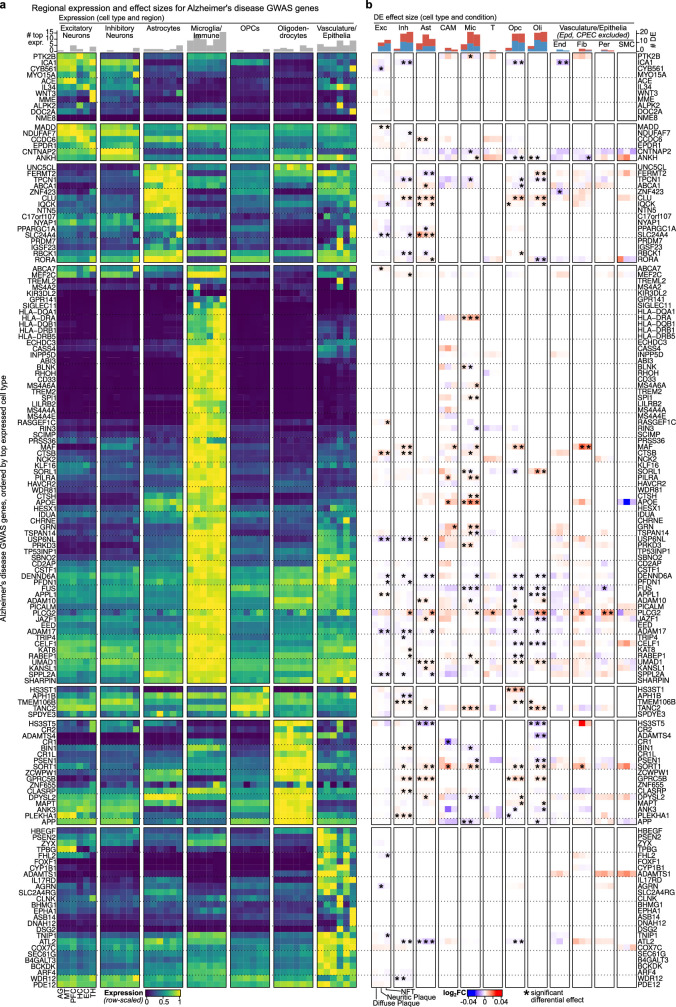
Alzheimer's disease is the leading cause of dementia worldwide, but the cellular pathways that underlie its pathological progression across brain regions remain poorly understood1-3. Here we report a single-cell transcriptomic atlas of six different brain regions in the aged human brain, covering 1.3 million cells from 283 post-mortem human brain samples across 48 individuals with and without Alzheimer's disease. We identify 76 cell types, including region-specific subtypes of astrocytes and excitatory neurons and an inhibitory interneuron population unique to the thalamus and distinct from canonical inhibitory subclasses. We identify vulnerable populations of excitatory and inhibitory neurons that are depleted in specific brain regions in Alzheimer's disease, and provide evidence that the Reelin signalling pathway is involved in modulating the vulnerability of these neurons. We develop a scalable method for discovering gene modules, which we use to identify cell-type-specific and region-specific modules that are altered in Alzheimer's disease and to annotate transcriptomic differences associated with diverse pathological variables. We identify an astrocyte program that is associated with cognitive resilience to Alzheimer's disease pathology, tying choline metabolism and polyamine biosynthesis in astrocytes to preserved cognitive function late in life. Together, our study develops a regional atlas of the ageing human brain and provides insights into cellular vulnerability, response and resilience to Alzheimer's disease pathology.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



Comment in
-
Single-cell atlas of Alzheimer disease vulnerability.Nat Rev Neurol. 2024 Sep;20(9):505. doi: 10.1038/s41582-024-01008-z. Nat Rev Neurol. 2024. PMID: 39107436 No abstract available.
References
MeSH terms
Substances
Grants and funding
- P30 AG072975/AG/NIA NIH HHS/United States
- R01 AG074003/AG/NIA NIH HHS/United States
- RF1 AG062377/AG/NIA NIH HHS/United States
- T32 GM087237/GM/NIGMS NIH HHS/United States
- P30 AG010161/AG/NIA NIH HHS/United States
- R01 NS127187/NS/NINDS NIH HHS/United States
- RF1 AG054321/AG/NIA NIH HHS/United States
- U01 NS110453/NS/NINDS NIH HHS/United States
- R01 AG062335/AG/NIA NIH HHS/United States
- U01 MH119509/MH/NIMH NIH HHS/United States
- R01 AG015819/AG/NIA NIH HHS/United States
- RF1 AG054012/AG/NIA NIH HHS/United States
- U01 AG046152/AG/NIA NIH HHS/United States
- R01 AG058002/AG/NIA NIH HHS/United States
- R01 HG008155/HG/NHGRI NIH HHS/United States
- U01 AG061356/AG/NIA NIH HHS/United States
- UG3 NS115064/NS/NINDS NIH HHS/United States
- R01 AG017917/AG/NIA NIH HHS/United States
- UH3 NS115064/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
