

Cryo-EM architecture of a near-native stretch-sensitive membrane microdomain
- PMID: 39048819
- PMCID: PMC11324527
- DOI: 10.1038/s41586-024-07720-6
Cryo-EM architecture of a near-native stretch-sensitive membrane microdomain
Abstract
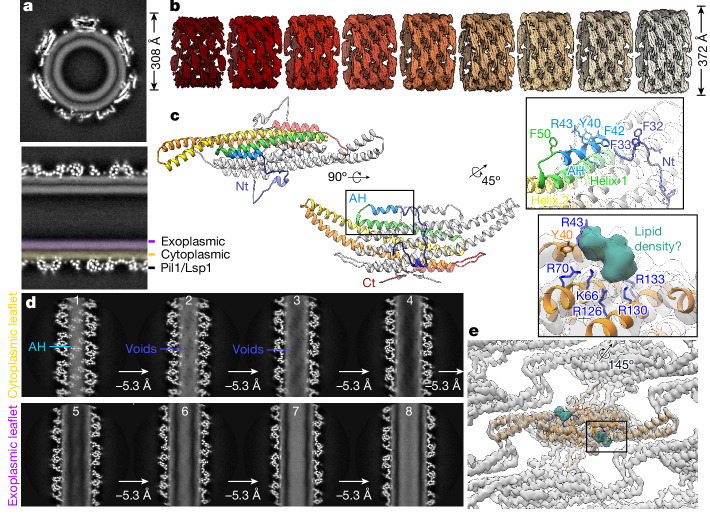
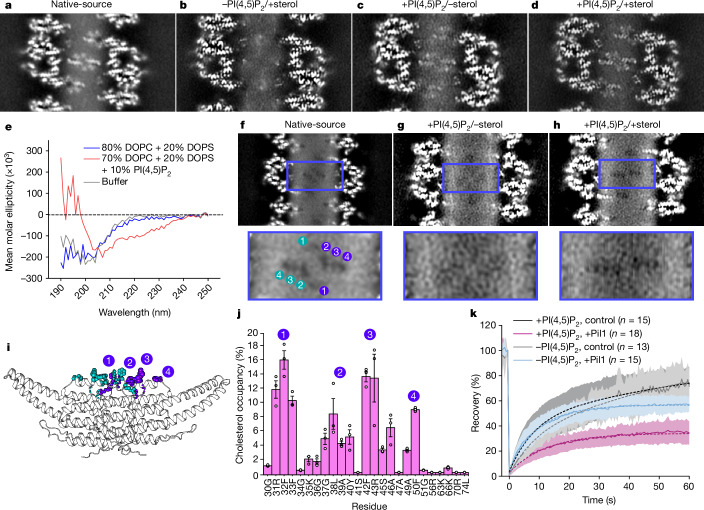
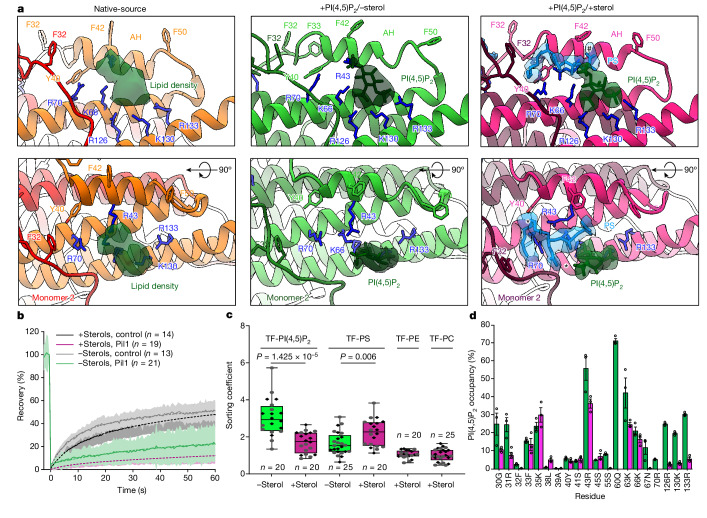
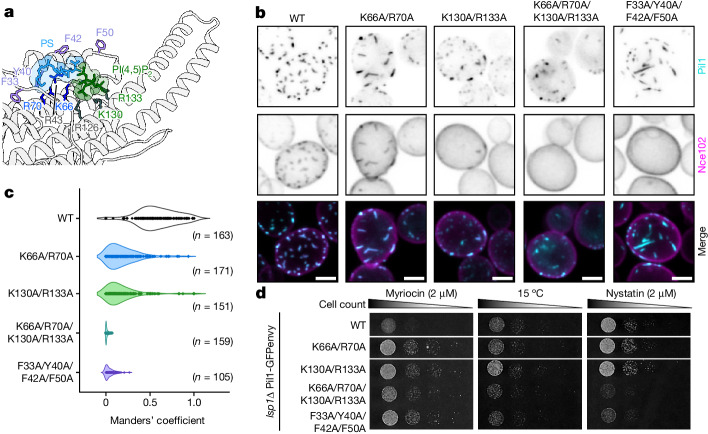
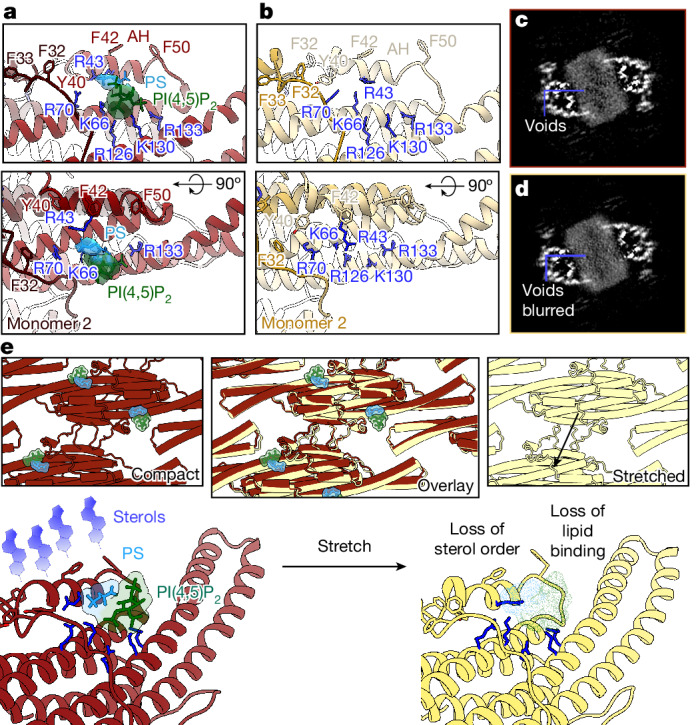
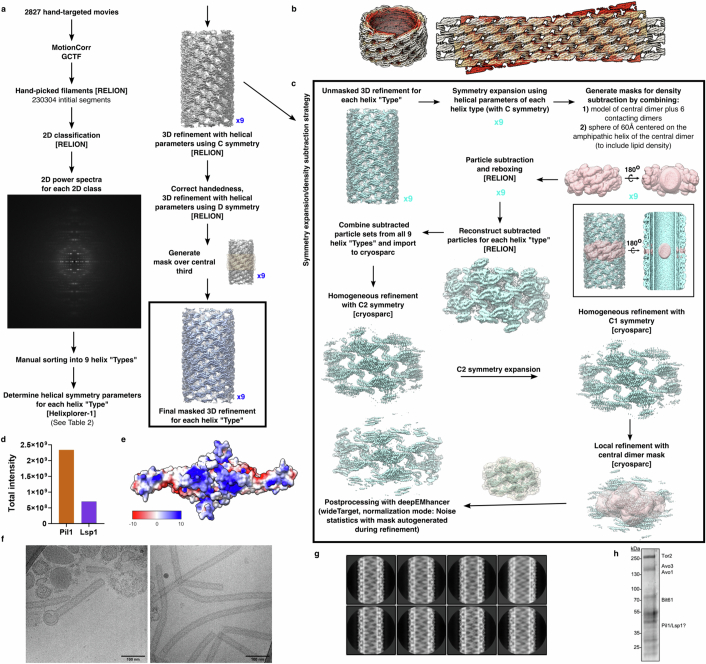
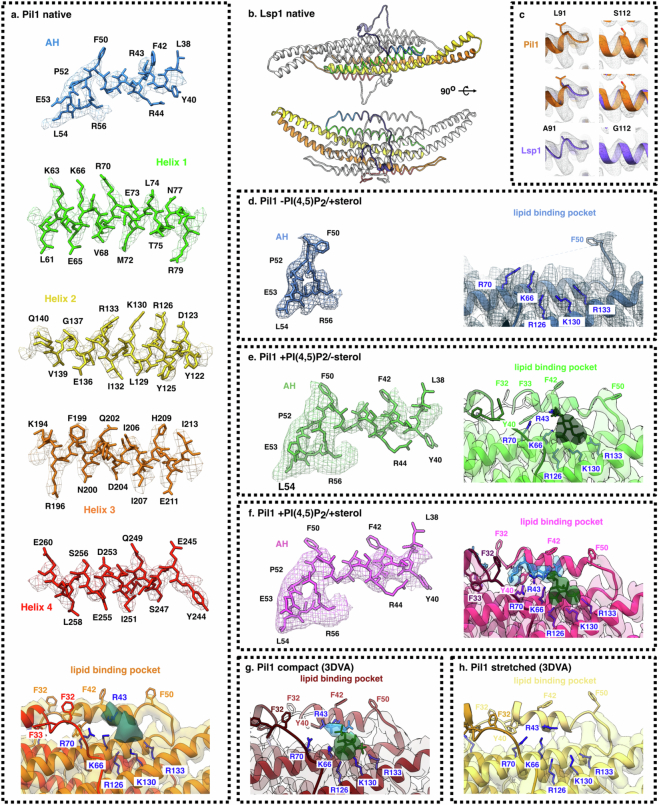
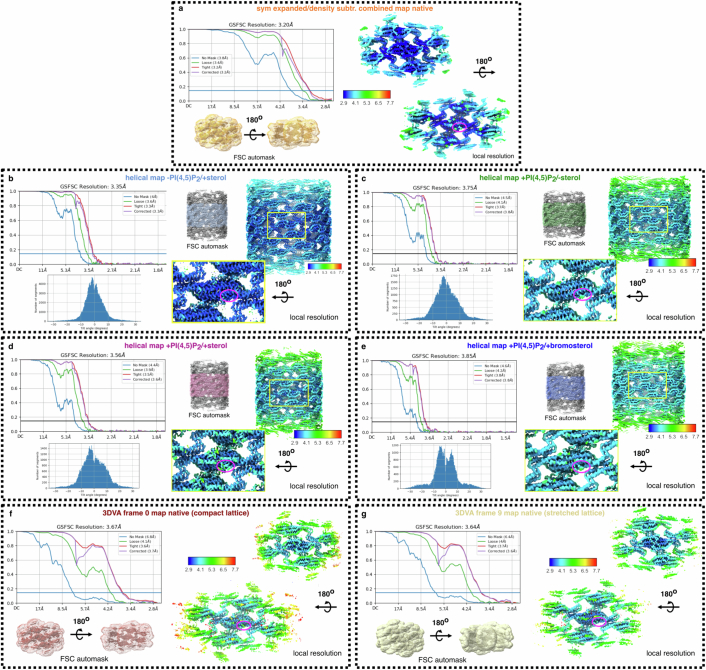
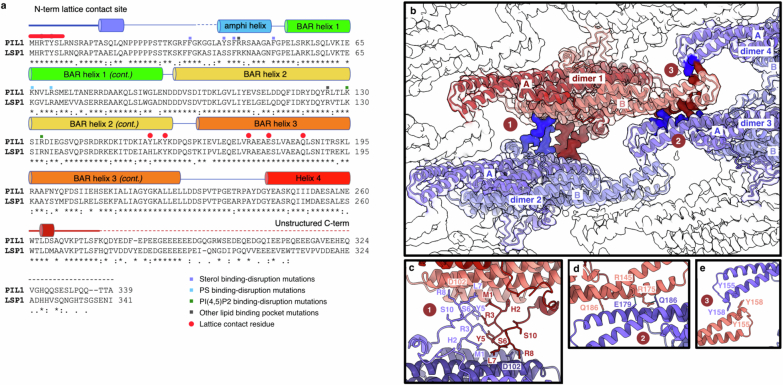
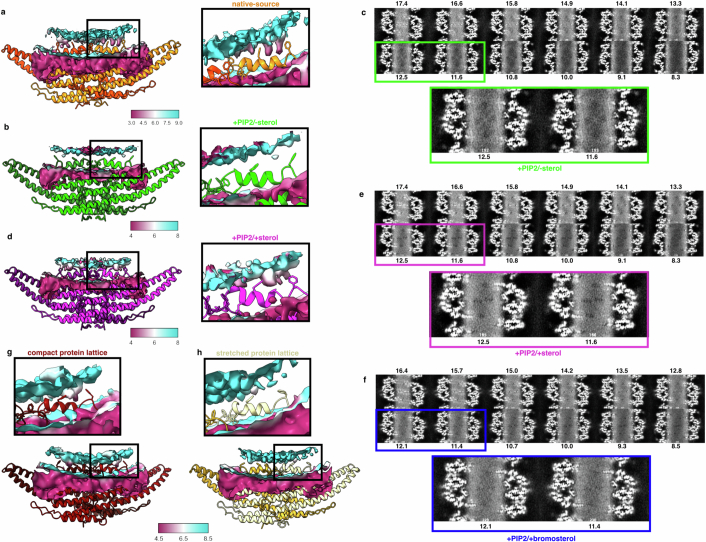
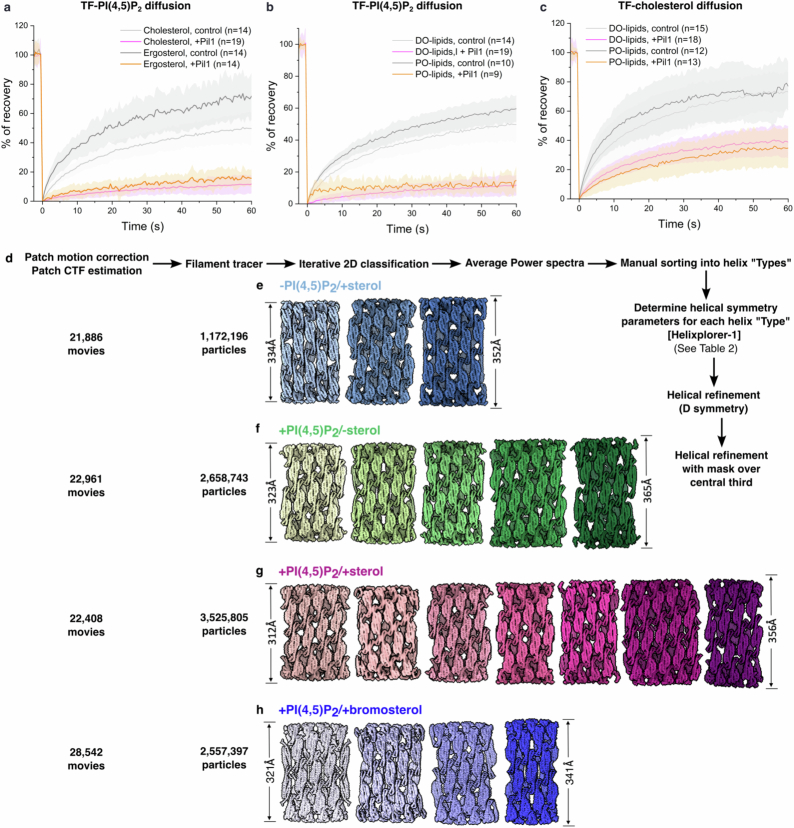
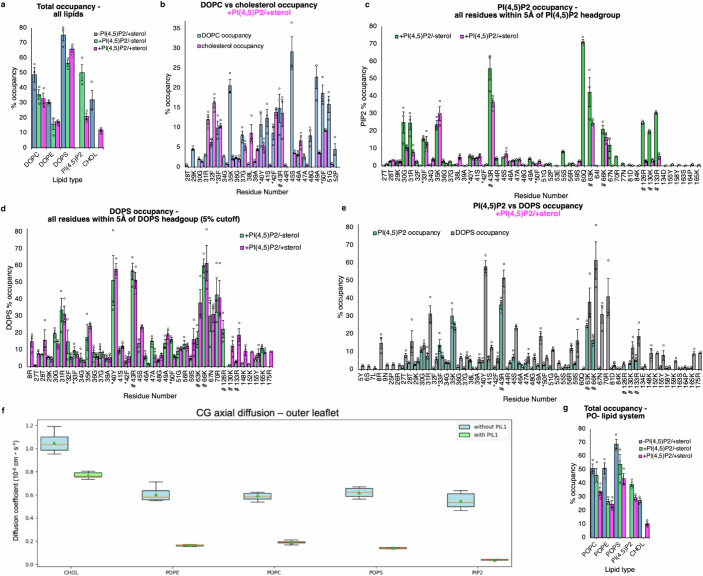
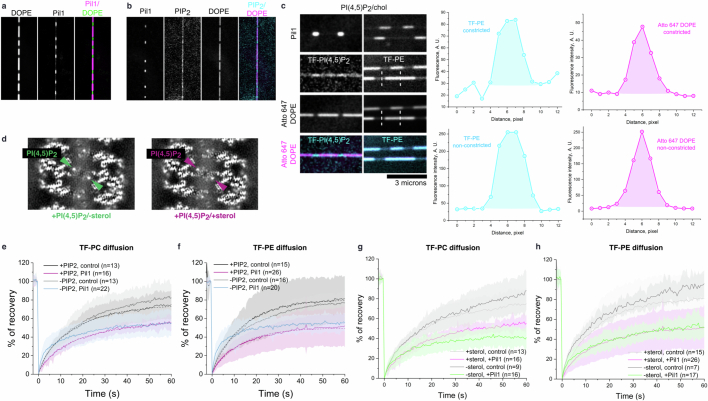
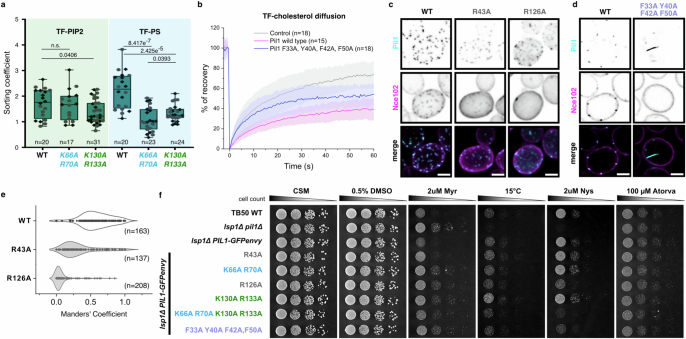
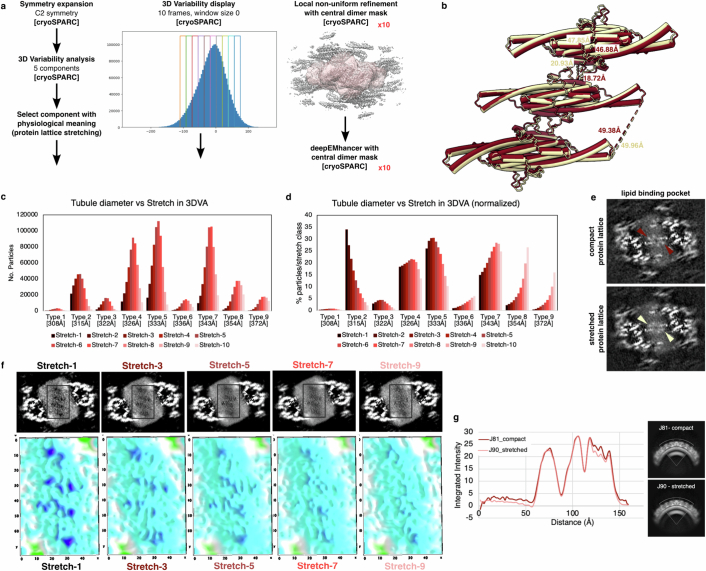
Biological membranes are partitioned into functional zones termed membrane microdomains, which contain specific lipids and proteins1-3. The composition and organization of membrane microdomains remain controversial because few techniques are available that allow the visualization of lipids in situ without disrupting their native behaviour3,4. The yeast eisosome, composed of the BAR-domain proteins Pil1 and Lsp1 (hereafter, Pil1/Lsp1), scaffolds a membrane compartment that senses and responds to mechanical stress by flattening and releasing sequestered factors5-9. Here we isolated near-native eisosomes as helical tubules made up of a lattice of Pil1/Lsp1 bound to plasma membrane lipids, and solved their structures by helical reconstruction. Our structures reveal a striking organization of membrane lipids, and, using in vitro reconstitutions and molecular dynamics simulations, we confirmed the positioning of individual PI(4,5)P2, phosphatidylserine and sterol molecules sequestered beneath the Pil1/Lsp1 coat. Three-dimensional variability analysis of the native-source eisosomes revealed a dynamic stretching of the Pil1/Lsp1 lattice that affects the sequestration of these lipids. Collectively, our results support a mechanism in which stretching of the Pil1/Lsp1 lattice liberates lipids that would otherwise be anchored by the Pil1/Lsp1 coat, and thus provide mechanistic insight into how eisosome BAR-domain proteins create a mechanosensitive membrane microdomain.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
