

Spinocerebellar Ataxias: Phenotypic Spectrum of PolyQ versus Non-Repeat Expansion Forms
- PMID: 39048885
- PMCID: PMC11585503
- DOI: 10.1007/s12311-024-01723-9
Spinocerebellar Ataxias: Phenotypic Spectrum of PolyQ versus Non-Repeat Expansion Forms
Abstract
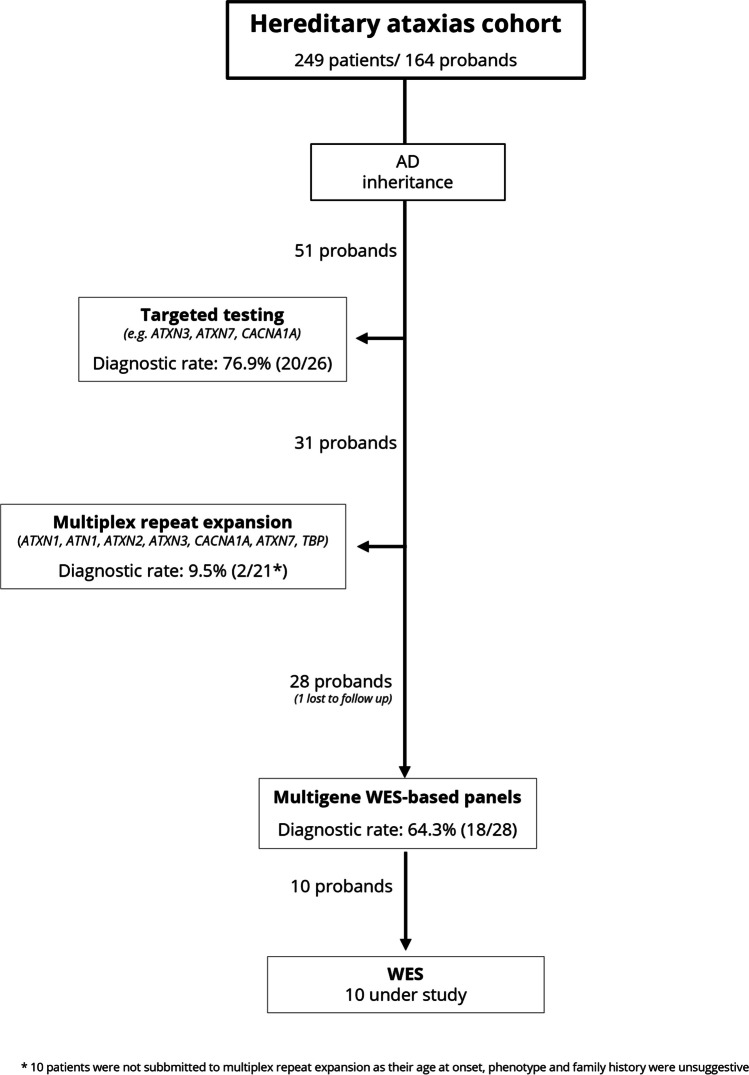
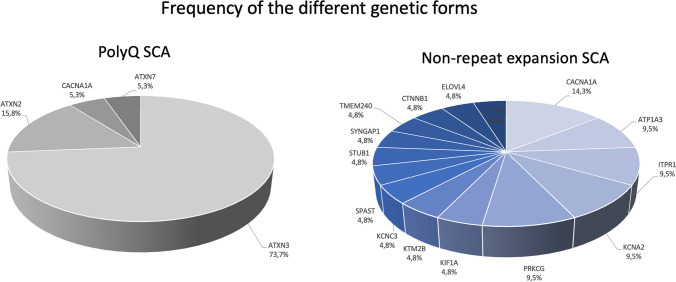
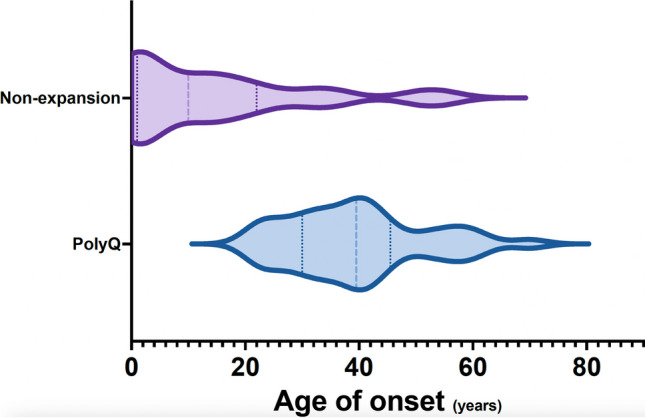
Spinocerebellar ataxias (SCA) are most frequently due to (CAG)n (coding for polyglutamine, polyQ) expansions and, less so, to expansion of other oligonucleotide repeats (non-polyQ) or other type of variants (non-repeat expansion SCA). In this study we compared polyQ and non-repeat expansion SCA, in a cohort of patients with hereditary ataxia followed at a tertiary hospital. From a prospective study, 88 patients (51 families) with SCA were selected, 74 (40 families) of whom genetically diagnosed. Thirty-eight patients (51.4%, 19 families) were confirmed as having a polyQ (no other repeat-expansions were identified) and 36 (48.6%, 21 families) a non-repeat expansion SCA. Median age-at-onset was 39.5 [30.0-45.5] for polyQ and 7.0 years [1.00-21.50] for non-repeat expansion SCA. PolyQ SCA were associated with cerebellar onset, and non-repeat expansion forms with non-cerebellar onset. Time to diagnosis was longer for non-repeat expansion SCA. The most common polyQ SCA were Machado-Joseph disease (MJD/SCA3) (73.7%) and SCA2 (15.8%); whereas in non-repeat expansion SCA ATX-CACNA1A (14.3%), ATP1A3-related ataxia, ATX-ITPR1, ATX/HSP-KCNA2, and ATX-PRKCG (9.5% each) predominated. Disease duration (up to inclusion) was significantly higher in non-repeat expansion SCA, but the difference in SARA score was not statistically significant. Cerebellar peduncles and pons atrophy were more common in polyQ ataxias, as was axonal neuropathy. SCA had a wide range of genetic etiology, age-at-onset and presentation. Proportion of polyQ and non-repeat expansion SCA was similar; the latter had a higher genetic heterogeneity. While polyQ ataxias were typically linked to cerebellar onset in adulthood, non-repeat expansion forms associated with early onset and non-cerebellar presentations.
Keywords: Hereditary Ataxia; Hereditary Spinocerebellar Degenerations; Machado-Joseph disease; Spinocerebellar Diseases; Trinucleotide Repeat Expansions.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethical Approval and Informed Consent: The authors confirm this work has been approved by institutional review board of ULSSA, and every patient gave written informed consent to participate in the project and for publication of the study’s results. The study was performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki, its later amendments or comparable ethical standards. Competing Interests: The authors declare no competing interests.
Figures
References
-
- Schöls L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3(5):291–304. - PubMed
-
- Klockgether T. Update on degenerative ataxias. Curr Opin Neurol. 2011;24(4):339–45. - PubMed
-
- Parodi L, Coarelli G, Stevanin G, Brice A, Durr A. Hereditary ataxias and paraparesias: clinical and genetic update. Curr Opin Neurol. 2018;31(4):462–71. - PubMed
-
- Sequeiros J, Coutinho P. Epidemiology and clinical aspects of Machado-Joseph disease. Adv Neurol. 1993;61:139–53. - PubMed
-
- Ashizawa T, Figueroa PK, Perlman LS, Gomez MC, Wilmot RG, Schmahmann DJ, Ying HS, Zesiewicz AT, Paulson LH, Shakkottai GV, Bushara OK, Kuo S-H, Geschwind DM, Xia G, Mazzoni P, Krischer PJ, Cuthbertson D, Holbert A, Ferguson HJ, et al. Clinical characteristics of patients with spinocerebellar ataxias 1, 2, 3 and 6 in the US; a prospective observational study. Orphanet J Rare Dis. 2013;8(1):177. - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
