

Vitamin K2 sensitizes the efficacy of venetoclax in acute myeloid leukemia by targeting the NOXA-MCL-1 pathway
- PMID: 39052583
- PMCID: PMC11271855
- DOI: 10.1371/journal.pone.0307662
Vitamin K2 sensitizes the efficacy of venetoclax in acute myeloid leukemia by targeting the NOXA-MCL-1 pathway
Abstract
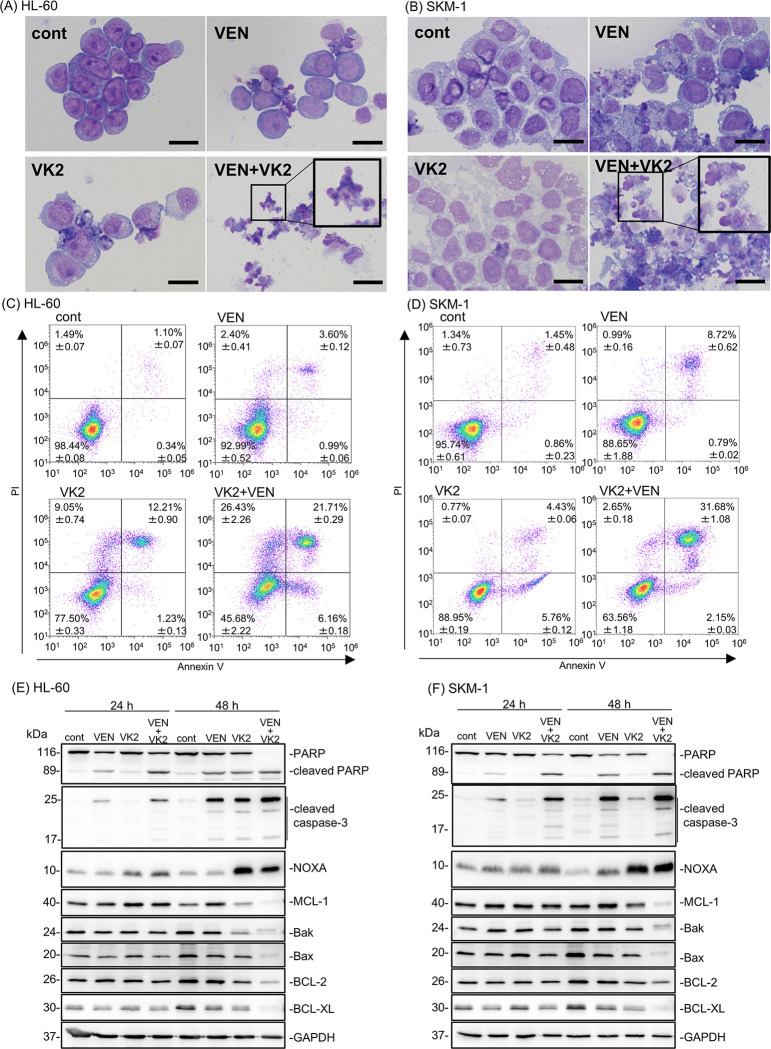
Promising outcomes have been reported in elder patients with acute myeloid leukemia (AML) using combined therapy of venetoclax (VEN) and azacytidine (AZA) in recent years. However, approximately one-third of patients appear to be refractory to this therapy. Vitamin K2 (VK2) shows apoptosis-inducing activity in AML cells, and daily oral VK2 (menaquinone-4, GlakayR) has been approved for patients with osteoporosis in Japan. We observed a high response rate to AZA plus VEN therapy, with no 8-week mortality in the newly diagnosed AML patients consuming daily VK2 in our hospital. The median age of the patients was 75.9 years (range 66-84) with high-risk features. Patients received AZA 75 mg/m2 on D1-7, VEN 400 mg on D1-28, and daily VK2 45 mg. The CR/CRi ratio was 94.7% (18/19), with a CR rate of 79%. Complete cytogenetic CR was achieved in 15 of 19 (79%) patients, and MRD negativity in 2 of 15 (13%) evaluable CR patients. Owing to the extremely high response rate in clinical settings, we further attempted to investigate the underlying mechanisms. The combination of VK2 and VEN synergistically induced apoptosis in all five AML cell lines tested. VK2, but not VEN, induced mitochondrial reactive oxygen species (ROS), leading to the transcriptional upregulation of NOXA, followed by MCL-1 repression. ROS scavengers repressed VK2 induced-NOXA expression and led to the cancellation of pronounced apoptosis and the downregulation of MCL-1 by VK2 plus VEN. Additionally, knockdown and knockout of NOXA resulted in abrogation of the MCL-1 repression as well as enhanced cytotoxicity by the two-drug combination, indicating that VK2 suppresses MCL-1 via ROS-mediated NOXA induction. These data suggest that the dual inhibition of BCL-2 by VEN and MCL-1 by VK2 is responsible for the remarkable clinical outcomes in our patients. Therefore, large-scale clinical trials are required.
Copyright: © 2024 Tauchi et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures





References
-
- Daver NG, Maiti A, Kadia TM, Vyas P, Majeti R, Wei AH, et al.. TP53-Mutated Myelodysplastic Syndrome and Acute Myeloid Leukemia: Biology, Current Therapy, and Future Directions. Cancer Discov. 2022; 12(11): 2516–2529. doi: 10.1158/2159-8290.CD-22-0332 . - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
