

Impaired B-cell function in ERCC2 deficiency
- PMID: 39055713
- PMCID: PMC11269123
- DOI: 10.3389/fimmu.2024.1423141
Impaired B-cell function in ERCC2 deficiency
Abstract
Background: Trichothiodystrophy-1 (TTD1) is an autosomal-recessive disease and caused by mutations in ERCC2, a gene coding for a subunit of the TFIIH transcription and nucleotide-excision repair (NER) factor. In almost half of these patients infectious susceptibility has been reported but the underlying molecular mechanism leading to immunodeficiency is largely unknown.
Objective: The aim of this study was to perform extended molecular and immunological phenotyping in patients suffering from TTD1.
Methods: Cellular immune phenotype was investigated using multicolor flow cytometry. DNA repair efficiency was evaluated in UV-irradiation assays. Furthermore, early BCR activation events and proliferation of TTD1 lymphocytes following DNA damage induction was tested. In addition, we performed differential gene expression analysis in peripheral lymphocytes of TTD1 patients.
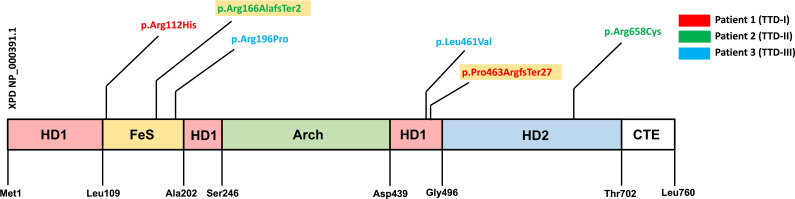
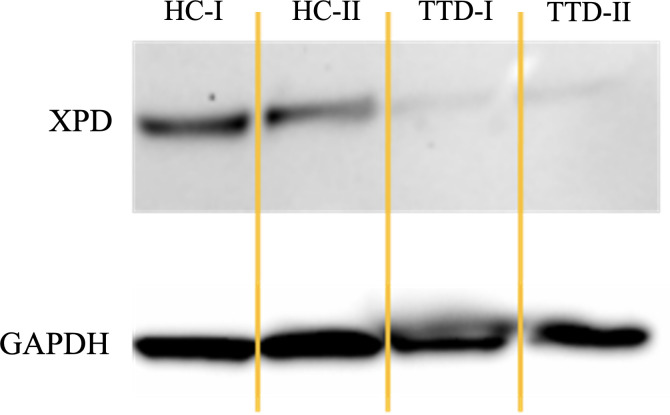
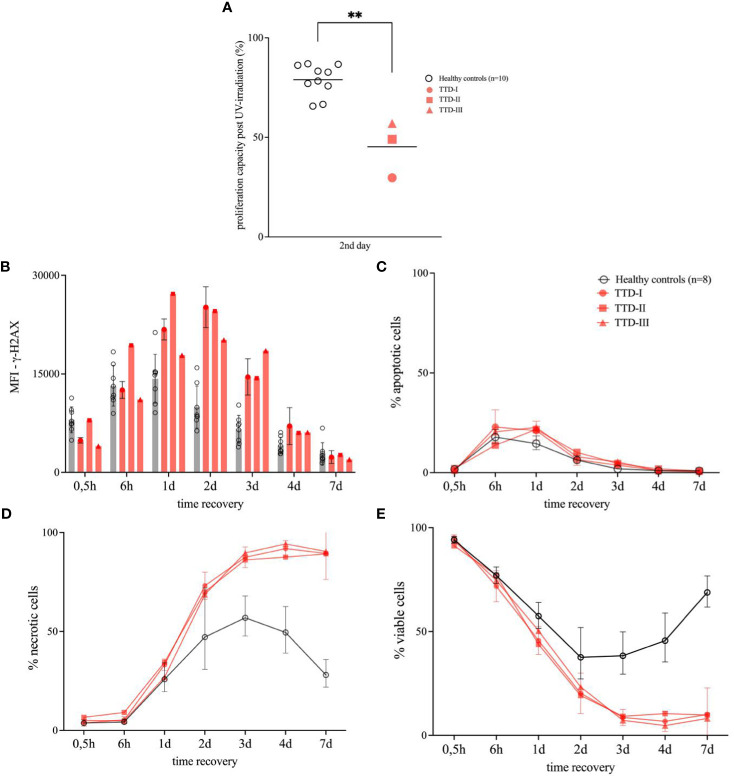
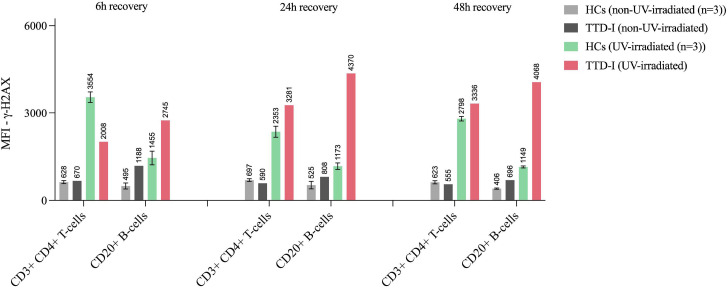
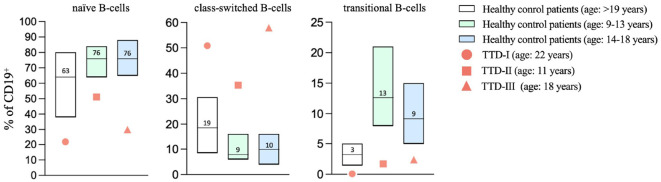
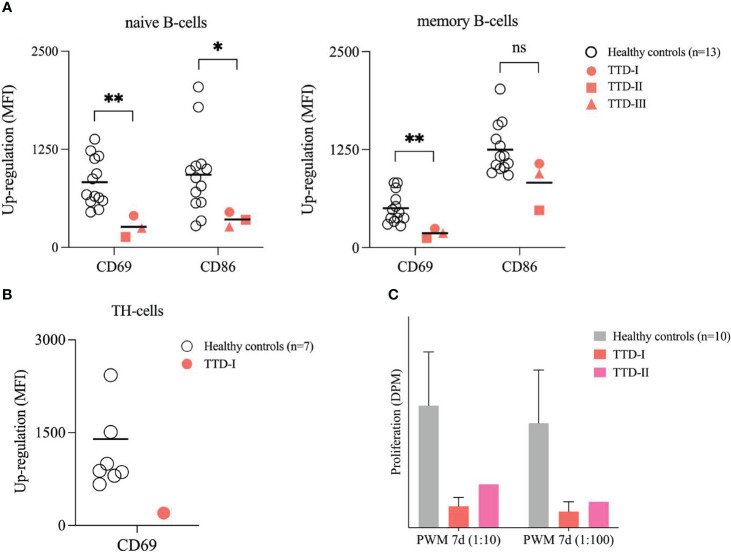
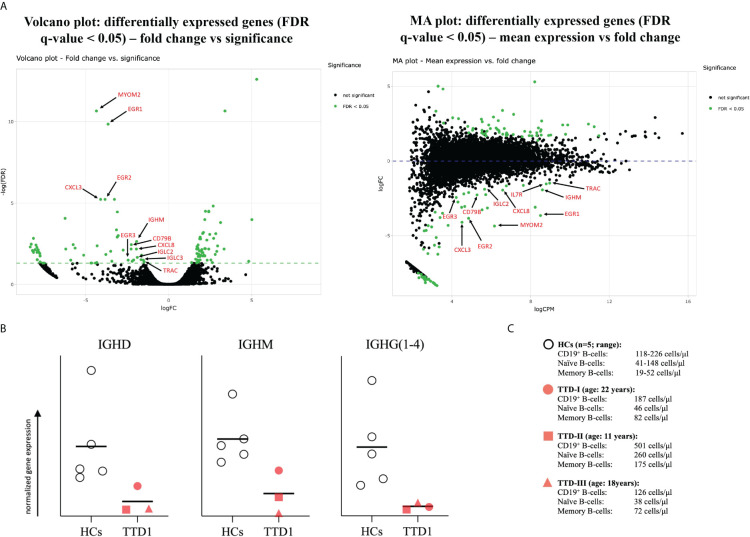
Results: We investigated three unrelated TTD1 patients who presented with recurrent infections early in life of whom two harbored novel ERCC2 mutations and the third patient is a carrier of previously described pathogenic ERCC2 mutations. Hypogammaglobulinemia and decreased antibody responses following vaccination were found. TTD1 B-cells showed accumulation of γ-H2AX levels, decreased proliferation activity and reduced cell viability following UV-irradiation. mRNA sequencing analysis revealed significantly downregulated genes needed for B-cell development and activation. Analysis of B-cell subpopulations showed low numbers of naïve and transitional B-cells in TTD1 patients, indicating abnormal B-cell differentiation in vivo.
Conclusion: In summary, our analyses confirmed the pathogenicity of novel ERCC2 mutations and show that ERCC2 deficiency is associated with antibody deficiency most likely due to altered B-cell differentiation resulting from impaired BCR-mediated B-cell activation and activation-induced gene transcription.
Keywords: B-cell activation; DNA repair deficiency; ERCC2; XPD; antibody deficiency; nucleotide excision repair; primary immunodeficiency; trichothiodystrophy.
Copyright © 2024 Rossmanith, Sauerwein, Geier, Leiss-Piller, Stemberger, Sharapova, Gruber, Bergler, Verbsky, Csomos, Walter and Wolf.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials