

Sodium-Glucose Cotransporter Inhibitors: Cellular Mechanisms Involved in the Lipid Metabolism and the Treatment of Chronic Kidney Disease Associated with Metabolic Syndrome
- PMID: 39061837
- PMCID: PMC11274291
- DOI: 10.3390/antiox13070768
Sodium-Glucose Cotransporter Inhibitors: Cellular Mechanisms Involved in the Lipid Metabolism and the Treatment of Chronic Kidney Disease Associated with Metabolic Syndrome
Abstract
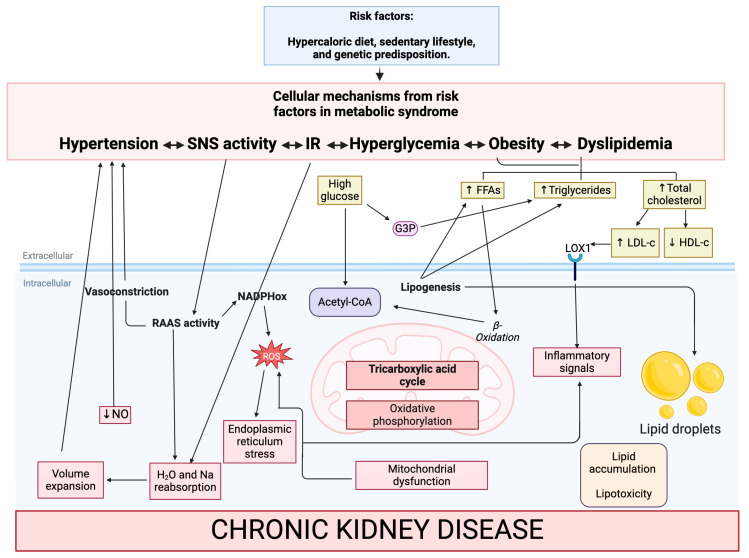
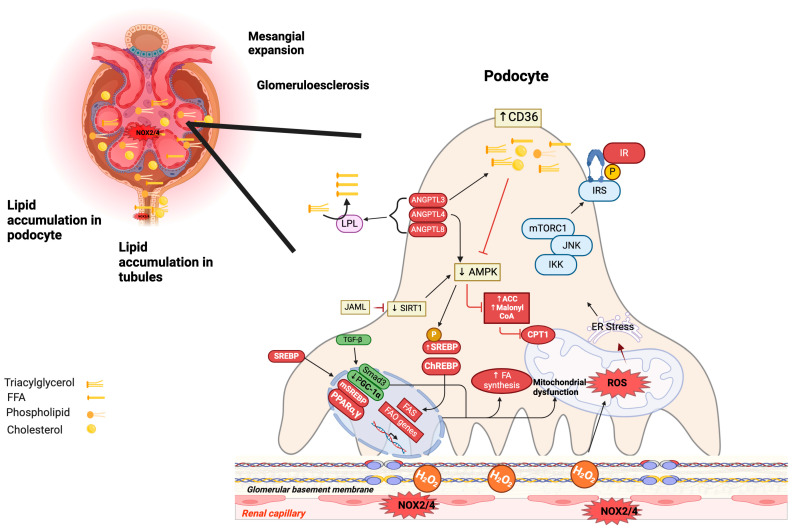
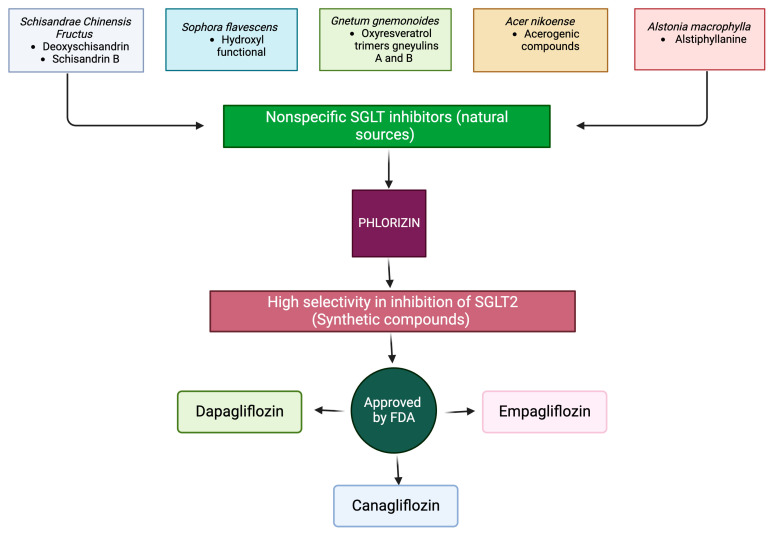
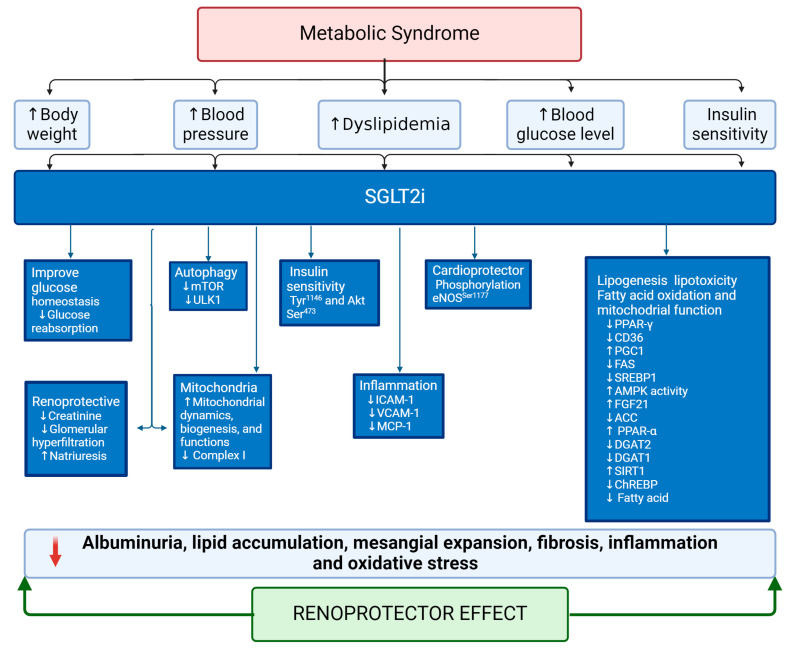
Metabolic syndrome (MetS) is a multifactorial condition that significantly increases the risk of cardiovascular disease and chronic kidney disease (CKD). Recent studies have emphasized the role of lipid dysregulation in activating cellular mechanisms that contribute to CKD progression in the context of MetS. Sodium-glucose cotransporter 2 inhibitors (SGLT2i) have demonstrated efficacy in improving various components of MetS, including obesity, dyslipidemia, and insulin resistance. While SGLT2i have shown cardioprotective benefits, the underlying cellular mechanisms in MetS and CKD remain poorly studied. Therefore, this review aims to elucidate the cellular mechanisms by which SGLT2i modulate lipid metabolism and their impact on insulin resistance, mitochondrial dysfunction, oxidative stress, and CKD progression. We also explore the potential benefits of combining SGLT2i with other antidiabetic drugs. By examining the beneficial effects, molecular targets, and cytoprotective mechanisms of both natural and synthetic SGLT2i, this review provides a comprehensive understanding of their therapeutic potential in managing MetS-induced CKD. The information presented here highlights the significance of SGLT2i in addressing the complex interplay between metabolic dysregulation, lipid metabolism dysfunction, and renal impairment, offering clinicians and researchers a valuable resource for developing improved treatment strategies and personalized approaches for patients with MetS and CKD.
Keywords: chronic kidney disease; dyslipidemia; hypertension; insulin resistance; lipid metabolism; lipotoxicity; metabolic syndrome; obesity; oxidative stress; sodium–glucose cotransporter 2 inhibitors.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Arellano Buendia A.S., Juárez Rojas J.G., García-Arroyo F., Aparicio Trejo O.E., Sánchez-Muñoz F., Argüello-García R., Sánchez-Lozada L.G., Bojalil R., Osorio-Alonso H. Antioxidant and anti-inflammatory effects of allicin in the kidney of an experimental model of metabolic syndrome. PeerJ. 2023;11:e16132. doi: 10.7717/peerj.16132. - DOI - PMC - PubMed
-
- Noubiap J.J., Nansseu J.R., Lontchi-Yimagou E., Nkeck J.R., Nyaga U.F., Ngouo A.T., Tounouga D.N., Tianyi F.L., Foka A.J., Ndoadoumgue A.L., et al. Geographic distribution of metabolic syndrome and its components in the general adult population: A meta-analysis of global data from 28 million individuals. Diabetes Res. Clin. Pract. 2022;188:109924. doi: 10.1016/j.diabres.2022.109924. - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
