

Nucleolytic processing of abasic sites underlies PARP inhibitor hypersensitivity in ALC1-deficient BRCA mutant cancer cells
- PMID: 39068174
- PMCID: PMC11283519
- DOI: 10.1038/s41467-024-50673-7
Nucleolytic processing of abasic sites underlies PARP inhibitor hypersensitivity in ALC1-deficient BRCA mutant cancer cells
Abstract
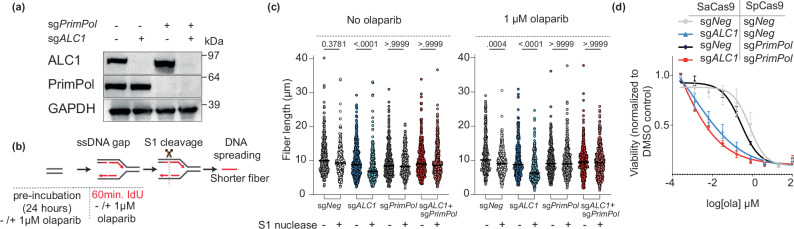
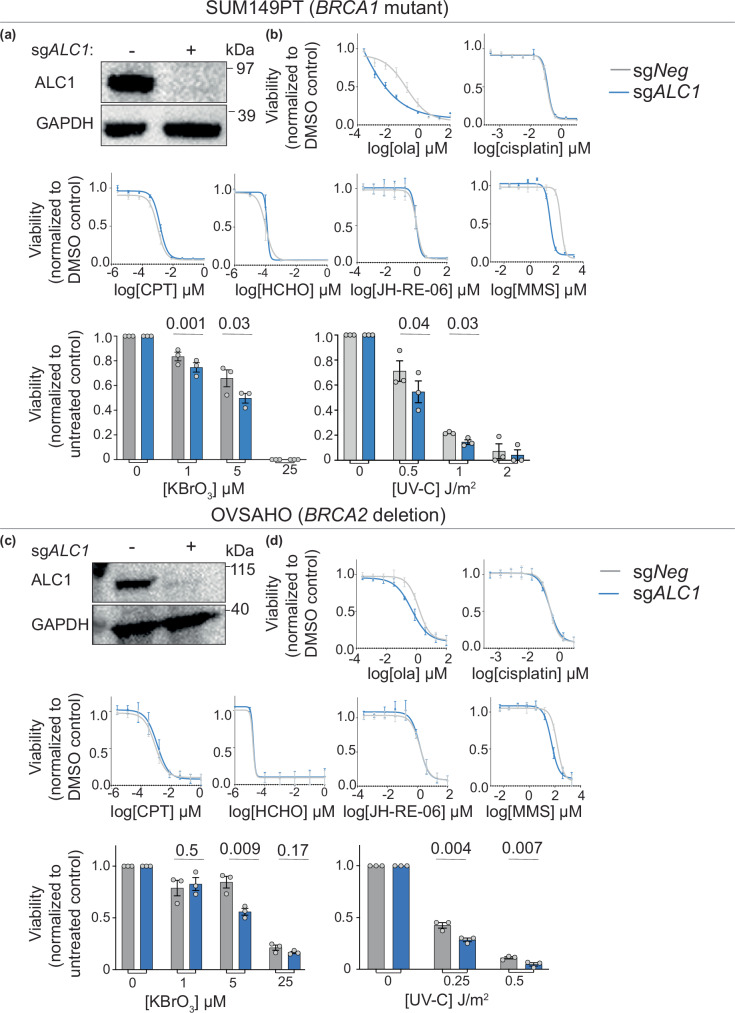
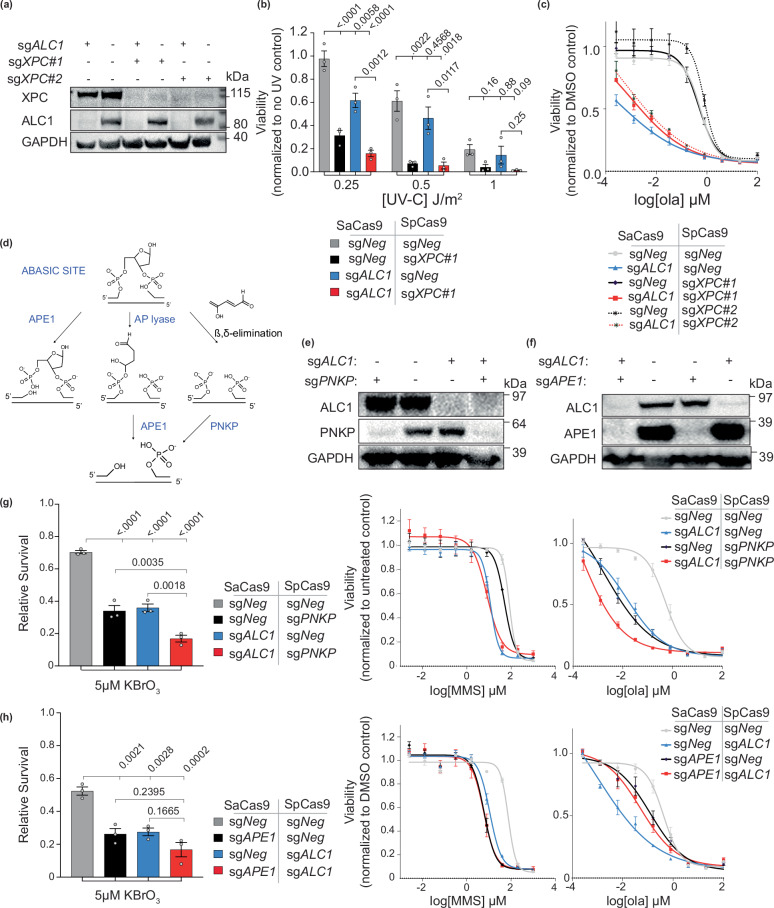
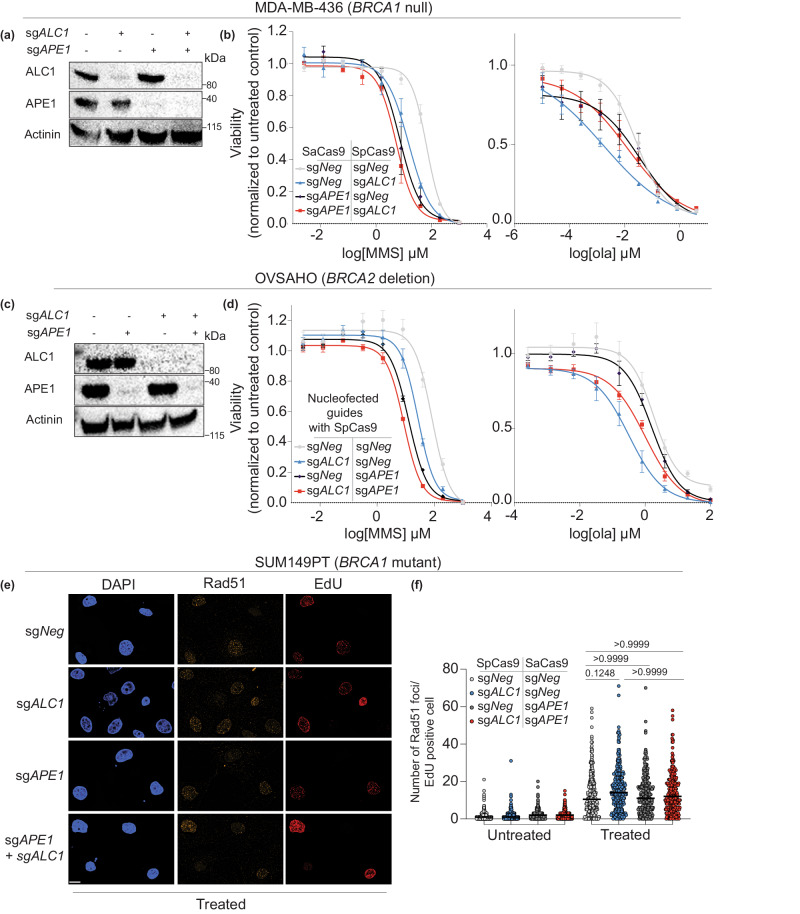
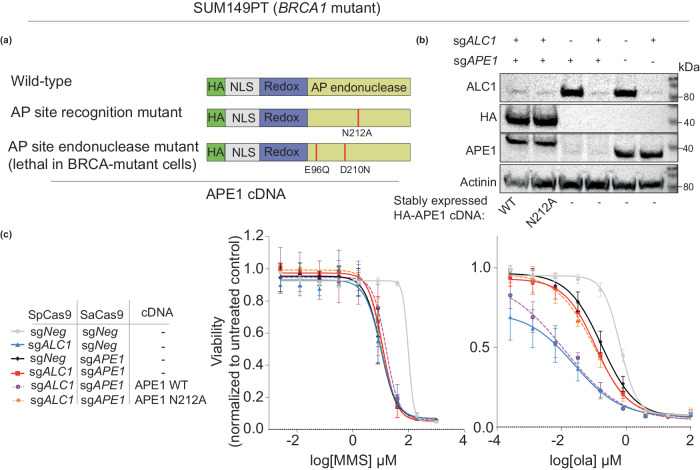
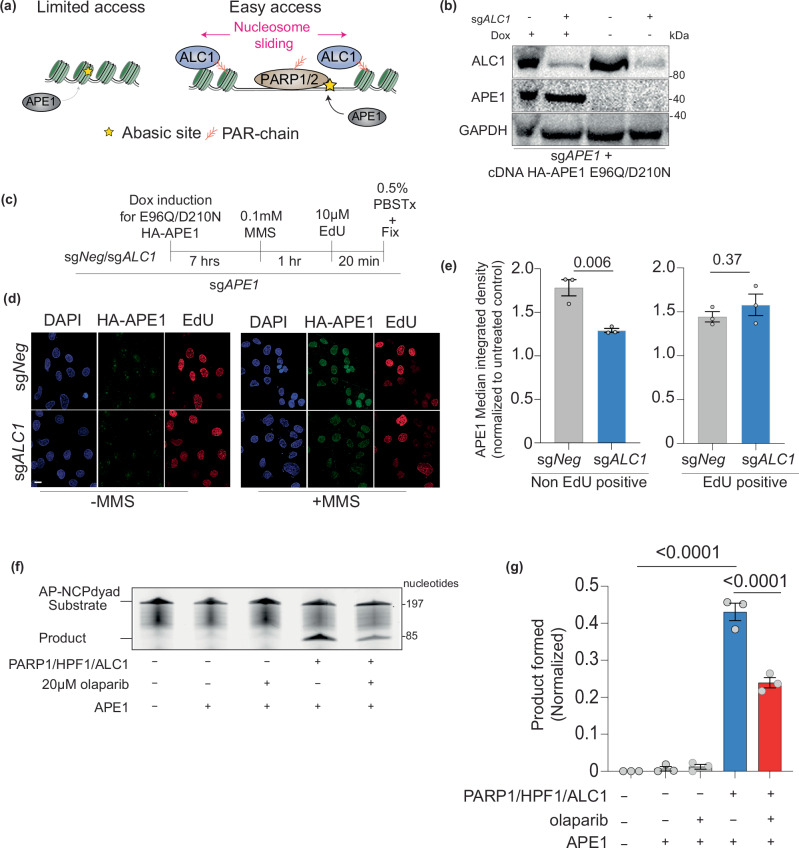
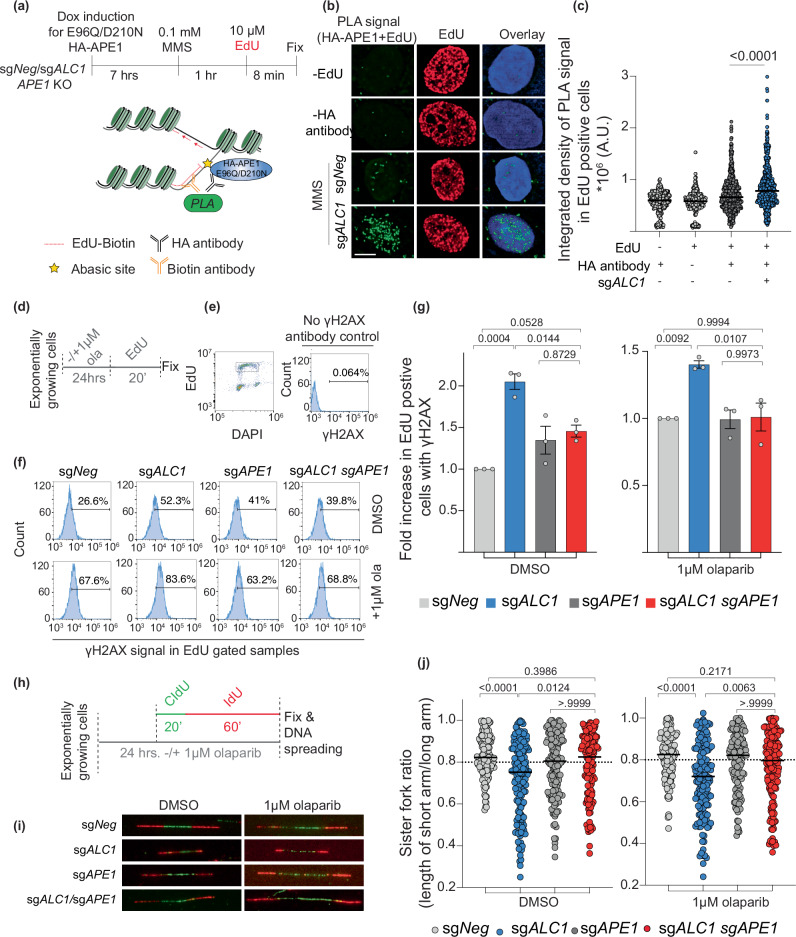
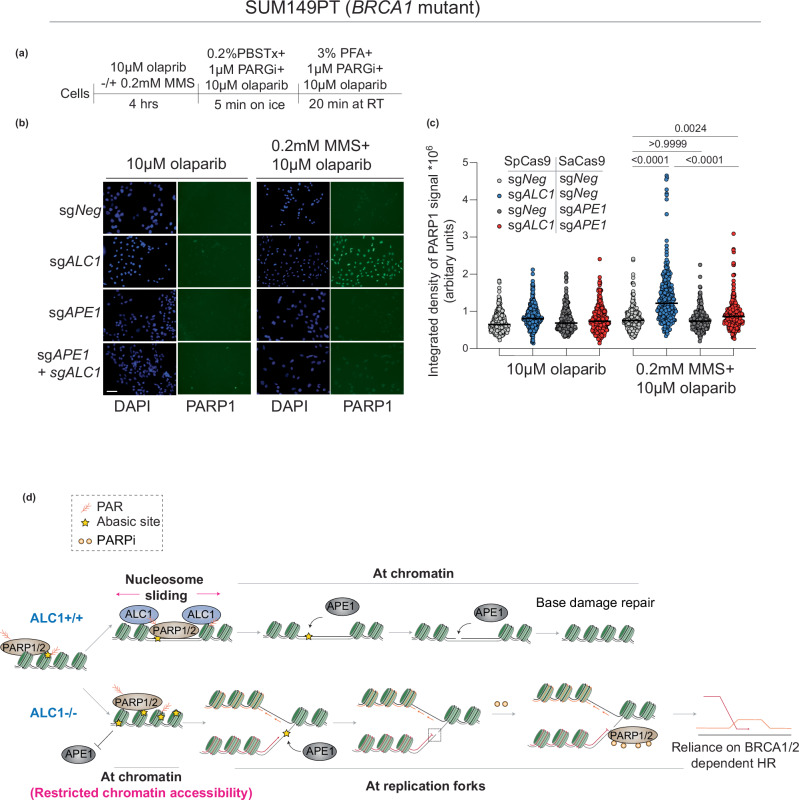
Clinical success with poly (ADP-ribose) polymerase inhibitors (PARPi) is impeded by inevitable resistance and associated cytotoxicity. Depletion of Amplified in Liver Cancer 1 (ALC1), a chromatin-remodeling enzyme, can overcome these limitations by hypersensitizing BReast CAncer genes 1/2 (BRCA1/2) mutant cells to PARPi. Here, we demonstrate that PARPi hypersensitivity upon ALC1 loss is reliant on its role in promoting the repair of chromatin buried abasic sites. We show that ALC1 enhances the ability of the abasic site processing enzyme, Apurinic/Apyrimidinic endonuclease 1 (APE1) to cleave nucleosome-occluded abasic sites. However, unrepaired abasic sites in ALC1-deficient cells are readily accessed by APE1 at the nucleosome-free replication forks. APE1 cleavage leads to fork breakage and trapping of PARP1/2 upon PARPi treatment, resulting in hypersensitivity. Collectively, our studies reveal how cells overcome the chromatin barrier to repair abasic lesions and uncover cleavage of abasic sites as a mechanism to overcome limitations of PARPi.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
- 1022/Mary Kay Foundation (TMKF)
- R37 CA286908/CA/NCI NIH HHS/United States
- R01 ES029203/ES/NIEHS NIH HHS/United States
- IRG2113364/American Cancer Society (American Cancer Society, Inc.)
- T32 CA113275/CA/NCI NIH HHS/United States
- V2022-025/V Foundation for Cancer Research (V Foundation)
- HT9425310133/U.S. Department of Defense (United States Department of Defense)
- ECIG202331010/Ovarian Cancer Research Fund Alliance (Ovarian Cancer Research Fund Alliance, Inc.)
- R37-CA286908/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- R01 CA248526/CA/NCI NIH HHS/United States
- R35 GM128562/GM/NIGMS NIH HHS/United States
- R35-GM128562/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R01CA248526/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- 940194/Rivkin Center for Ovarian Cancer (Rivkin Center)
- R01 CA237263/CA/NCI NIH HHS/United States
- R01-ES029203/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- CCR231014077/Susan G. Komen (Susan G. Komen Breast Cancer Foundation)
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
